



Metabolic Model
“All Disease Begins in The Gut.”
- Hippocrates
Characterization of E. coli Nissle 1917
As a part of our project, we decided to thoroughly characterize the probiotic E. coli Nissle 1917 strain (Project / Nissle). To validate and strengthen our laboratory results, we chose genome scale metabolic modeling by conducting flux balance analysis.
This approach is based on thermodynamic processes. This means that a minimization of the Gibbs free energy is aspired.
$$\Delta G_r = \Delta G^o_r + R T \ln{Q}$$
Consequently, reactions within the organism need to be set as irreversible in the thermodynamically feasible direction. This leaves us with boundary conditions for the Gibbs free energy. In the study that was done about reconstructing metabolic models on a genome scale [3] two more assumptions were made:
- ATP-consuming reactions were not allowed to proceed in the reverse direction
- Reactions present in thermodynamically curated models assumed the reversibility constraints adopted in these models
These assumptions enabled us to use CarveMe to build a genome scale metabolic model of E. coli Nissle 1917, since all constraints were already given in a universal bacterial model, which could be used as a reference.
In our project, we chose E. coli Nissle 1917 because of its probiotic properties, which also confer to less overall reactions in taking place within the organism. This is also a crucial information for future teams willing to work with E. coli Nissle 1917. We confirmed this simplicity using escher maps built from an E. coli core metabolic model and from our E. coli Nissle 1917 metabolic model.

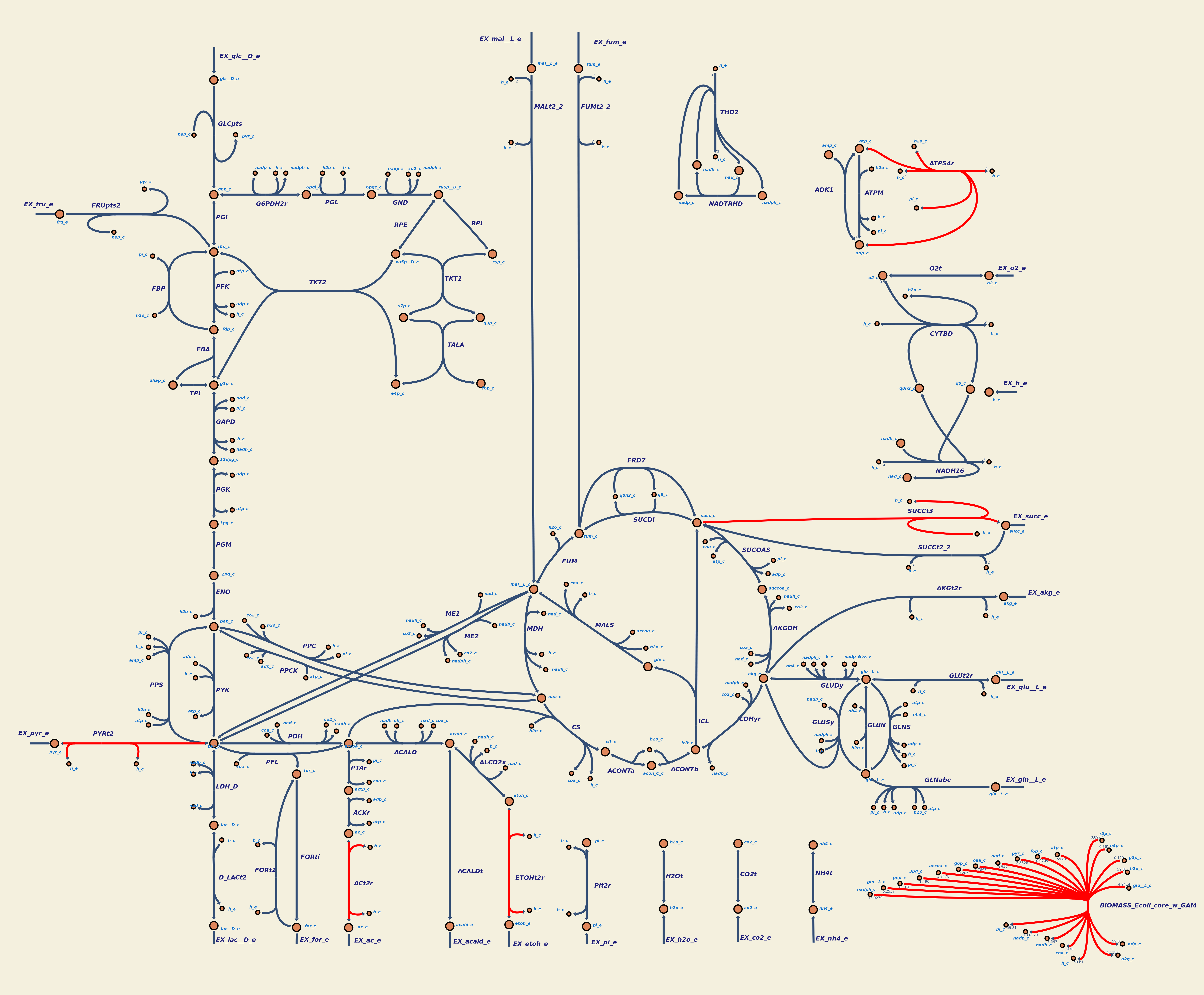
Additionally, two different media compositions were chosen for the simulation: Minimal medium M9, as well as complete (LB) medium, where all uptake reactions were allowed to carry flux, were chosen. Applying the minimal medium composition we identified the 2-Oxoglutarate exchange (EX_akg_e), the L-Glutamate exchange (EX_glu__L_e) and the D-tartrate exchange (EX_tartr__D_e) as limiting reactions within E. coli Nissle 1917 grown in a minimal medium (compare Figure 3).
Using the similar constraints, it also became apparent that the growth of EcN is strongly dependent on the O2 exchange (EX_o2_e) (compare Figure 4).
This led us to the comparison of anaerobic conditions with aerobic conditions. Optimizing the growth of E. coli Nissle 1917 on an aerobic medium resulted in a dimensionless growth rate of 99.919222 (Flux) with reduced costs of 2.220446e-16. In contrast, in an anaerobic medium, we yielded the same rate of 99.919222 for the flux, but the reduced costs changed to 1.096345e-15. This is in accordance with our wetlab results, which can be found under Project / Nissle: Basically there is a faster growth with oxygen since the strain has to first switch to fermentation under anaerobe conditions. As a consequence, we were aware of that in our lab experiments and calculated extra time for all anaerobe experiments we did.

Motivation of modeling the gut microbiome
Since our GMO is a probiotic acting in the patient’s gut, we wanted to go a step further and investigate the gut microbiome and its potential interactions with E. coli Nissle 1917. Despite the fact that clinical trials are not in the scope of the iGEM competition, we nevertheless decided that the gut microbiome will have a major influence on the behavior, and therefore the effectiveness, of our probiotic. This is why we also talked to Dr. Lisa Maier about possible influences of diseases on the human microbiome. Dr. Lisa Maier ( Human Practices / Experts) also hinted towards a possible experimental set-up to evaluate the interaction of EcN with the gut microbiome, using bacterial supernatant during E. coli Nissle 1917 growth experiments.
To gain further insights, we decided to model the behaviourojectr of three different gut microbiome compositions with respect to metabolic resource overlap (MRO), metabolic interaction potential (MIP), species coupling score (SCS), metabolic uptake score (MUS), metabolite production score (MPS) and a final SMETANA score integrating SCS, MUS and MPS. To our knowledge, such an analysis has never been done before within iGEM.
Building metabolic models for multiple bacteria
Since metabolic models for several of E. coli Nissle 1917’s competitors did not yet exist, we built them as described above. All models were created using protein genome files downloaded from the NCBI server [1] and are available to the scientific and the iGEM community under Parts / Downloads.
Choosing different microbiome compositions
Using all of our generated models, we grouped the bacteria into three different compositions. The first group of 5 organisms was the one we decided to use for our RNA-Seq experiments (see Project / Nissle): We chose Bacteroides thetaiotaomicron, Prevotella copri and Ruminococcus gnavus since they are commonly found within the human microbiome [4, 5]. Bifidobacterium adolescentis was chosen due to its probiotic nature and Clostridium difficile supernatant was used, since it is often found in the microbiome of people with chronic inflammation [4, 5].
The next composition of 19 organisms was chosen according to a very recent publication [6] covering : Anaerostipes hadrus, Bacteroides caccae, Bacteroides cellulosilyticus, Bacteroides dorei, Bacteroides ovatus, Bacteroides uniformis, Bacteroides vulgatus, Bacteroides xylanisolvens, Bifidobacterium adolescentis, Bifidobacterium longum, Blautia obeum, Collinsella aerofaciens, Dorea longicatena, Faecalibacterium prausnitzii, Parabacteroides distasonis, Parabacteroides merdae, Roseburia faecis and Escherichia coli. Two organisms were left out because no reliable genome sequence data could be obtained.
The third composition of 12 organisms was picked according to the "Todar's online textbook of bacteriology." [7]: Bacteroides fragilis, Bacteroides melaninogenicus, Bacteroides oralis, Clostridium perfringens, Clostridium septicum, Clostridium tetani, Bifidobacterium bifidum, Staphylococcus aureus, Enterococcus faecali, Escherichia coli and Salmonella enteritidis.
Discussion of different characteristics
We will only discuss the most meaningful results of the microbiome model in the following section. A download of the complete table of results is available under see Parts / Downloads.
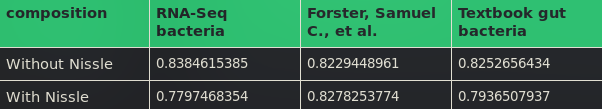
The metabolic resource overlap scores where calculated both with and without E. coli Nissle 1917 in the microbiome composition (Table 1). MRO is an intrinsic property of any community, independent of the habitat and robust in view of multiple minimal nutritional requirements for any (or all) member species, and providing an upper bound on the overall degree of resource competition [2]. It is computed using nutrient availabilities:
$$MRO = \frac{n \sum_{i,j, i \neq j} \left| M_i \cap M_j \right|}{C(n,2) \sum_{i=0}^N \left| M_i \right|}$$
Normally, communities consisting of phylogenetically closely related member species show a higher resource overlap than less related species.
According to the data denoted in Table 1, it is observable that E. coli Nissle 1917 most likely causes no resource threat to the gut microbiome since the MRO scores for the RNA-Seq composition (the bacteria from which the Bacterial supernatant was isolated) and the Textbook composition are getting lower. The score for the Forster, Samuel C., et al. composition almost stagnates.
The MIP is defined as the maximum number of essential nutritional components that a community can provide for itself through interspecies metabolic exchanges [2]. For our models it was not possible to compute MIP scores due to them being newly built from scratch up without subcommunity information.
The SMETANA score estimates the strength of metabolic coupling in the community through enumerating possible metabolite exchange is also denoted for every interaction in the complete table of results which is available under Parts / Downloads. It was calculated as the sum of all interspecies dependencies under a given nutritional environment. The growth dependency of species A on metabolite m produced by species B was calculated as a product of three separate scores: (i) species coupling score (SCS), (ii) metabolite uptake score (MUS), and (iii) metabolite production score (MPS). Dependency scores are normalized to range between 0 (complete independence) and 1 (essentiality) [2].
When comparing the results from working with SMETANA on all compositions without and with E. coli Nissle 1917, it is observable that E. coli Nissle 1917 changes the parameters which are important for all metabolic couplings throughout the metabolic community hence the gut microbiome. We denoted the highest dependencies of all compositions in the Tables 2 to 7.
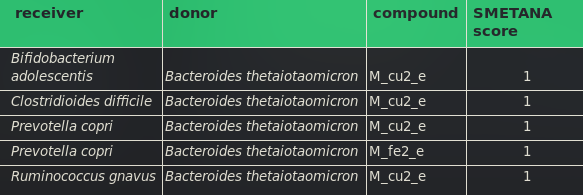




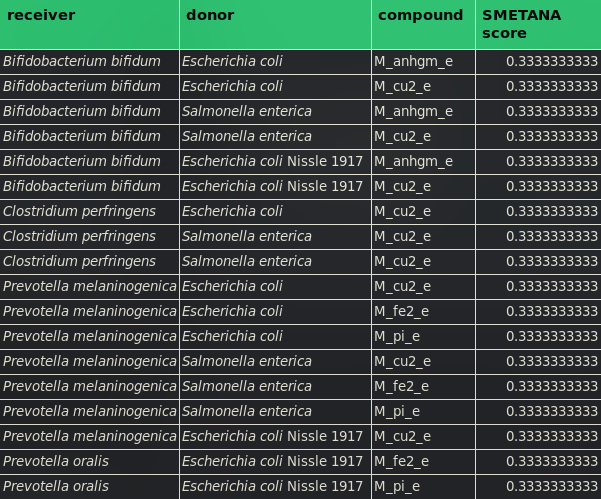
From the data gathered to fill the Tables above we can conclude that Fe2+ exchange (M_fe2_e), Cu2+ exchange (M_cu2_e), Fe3+ exchange (M_fe3_e) and Phosphate exchange (M_pi_e) have the biggest influence on the growth dependencies within the gut microbiome.
Through supplementation of those compounds/salts we could reduce the effect the introduced E.coli Nissle has on the gut Microbiome. In therapy this could be achieved through adding those compounds to the Pill or medium the Probiotic is given in, and through adjusting the diet towards an increased uptake of Iron and Copper.
Additional possibilities with metabolic models
Metabolic modelling and microbiome scale metabolic modelling are new, highly promising, cutting edge methods to gain further insight into bacterial behavior on all scales. With the orderly approach we presented here, we hope that future iGEMers can integrate these kind of models into their projects. For the completion of our project as described in Project / Outlook we plan to integrate the metabolic processes originating from the transgenes we add into the E. coli Nissle 1917 strain into our metabolic model. This will give us the opportunity to further deepen our knowledge of its interactions with the gut microbiome which is also a crucial part of turning this iGEM Project into a Start-Up as described in Project / Entrepreneurship. Metabolic modelling could also be the first step into designing synthetic microbial communities, which could become a new part of iGEM in the future.
RNA-Seq
Our extensive E. coli Nissle transcriptome examination, which is demonstrated on the Project / Nissle page is part of our modelling. All newly found overexpressed genes are possible candidates to overexpress in the laboratories to create more stress resistant E. coli strains, which are likelier to survive in the gut. Such approaches have previously been shown to work well [8].
For the application of our killswitch it will be interesting to check for possible regulators responding to cold stress. Such a system would be perfect to implement a cold switch killing the microbe after leaving the warmth of the body. This is part of our NCCT samples.
Hence, after iGEM, especially due to the high interest of several groups in Tübingen, we will continue our work on improving E. coli Nissle as a general probiotic. Our goal will be to establish more stress resistant strains
Cell Penetrating Peptides
To select the best CPP for the transport of Exendin-4, we generated a machine learning-based model, which predicts the transport efficiency of CPPs. The model was incorporated into our software-tool C3Pred to make our results publicly accessible for other teams.
Details of C3Pred can be found Project / Software.
We analyzed a series of CPP, which were already present in the iGEM Registry and additional peptides, which were frequently referred to in the literature.
Since Penetratin showed the highest scores compared to the other parts already available in the iGEM Registry, we decided to select this specific CPP as the carrier for the Exendin-4 protein in our project. Moreover, we decided to select Pentratin over other CPPs which had a higher activity score like pVEC, since Penetratin is frequently used in literature.
Additionally, we computed an elevated transport activity for the CPP fusion protein TAT-LK15, which has been reported to enhance the properties of TAT.
- TAT-LK15 : 228.81
References
- Website of the NCBI Genome server (https://www.ncbi.nlm.nih.gov/genome).
- Zelezniak, Aleksej, et al. "Metabolic dependencies drive species co-occurrence in diverse microbial communities." Proceedings of the National Academy of Sciences 112.20 (2015): 6449-6454. doi:/10.1073/pnas.1421834112
- Machado, Daniel, et al. "Fast automated reconstruction of genome-scale metabolic models for microbial species and communities." Nucleic acids research 46.15 (2018): 7542-7553. doi:/10.1093/nar/gky537
- Belizário JE, Napolitano M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front Microbiol. 2015;6:1050. Published 2015 Oct 6. doi:10.3389/fmicb.2015.01050
- Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med. 2016;8(1):51. Published 2016 Apr 27. doi:10.1186/s13073-016-0307-y
- Forster, Samuel C., et al. "A human gut bacterial genome and culture collection for improved metagenomic analyses." Nature biotechnology 37.2 (2019): 186. doi:/10.1038/s41587-018-0009-7
- Todar, Kenneth. "Todar's online textbook of bacteriology." (2006). , accessed October 2019
- Mills, Susan, et al. "Enhancing the stress responses of probiotics for a lifestyle from gut to product and back again." Microbial cell factories. Vol. 10. No. 1. BioMed Central, 2011.
- Visit icons8.com for the beautiful icons we used.

