

Description
The vision of accessible chassis-agnostic directed evolution

More than fifty years ago, a peculiar polymerase was isolated from the single-stranded RNA coliphage, Qβ.1 Around the same time thoughts of synthesizing evolution were emerging. The two events were not unrelated. In fact, the scientific histories of both directed evolution and Qβ replicase were entangled in the beginning, since Spiegelman’s Monster experiment.2 Today, Qβ’s RNA-inducible RNA polymerase is a well-characterized protein and directed evolution has won Arnold Francis’ 2018 Nobel Prize in chemistry for her contribution to the developing field of protein engineering. Despite these two subjects proximity, never has their relationship been exploited to realize the full potential of a directed evolution system.
The 2019 Stanford iGEM team saw an opportunity in the narrative above. Beginning our exploration for project ideas, we familiarize ourselves with the current hot-topic that is protein engineering via directed evolution. We researched many methodologies encompassing past iGEM projects like with PADE by Heidelberg’s 2017 team as well as more institutionalized methods such as OrthoRep by Chang Liu and PACE among many others. During our investigation of the topic, we noted the drawbacks that most methods share: time inefficiency, dependence upon the chassis organism, costs, and its subsequent inaccessibility.3 These difficulties that come with implementing directed evolutionary methods did not deter us; rather, we saw these disadvantages to current methodologies as an opportunity to improve such a foundational tool. Thus, we determined directed evolution to be the platform on which we will work. Our motivation for doing so is not only to offer another way of creating new proteins but also increase the accessibility to do so.
This is where Qβ comes in. This interesting replicase appeared to us as a potential solution to some common problems in directed evolution. Every method has its advantages and disadvantages, for instance, some have inherent biases in SNP formation. Others are non-specific in nature, creating SNPs throughout a vector or entire genome. But Qβ’s unique properties such as expressing no mutagenic biases for transversions to transitions nor for A/T and G/C substitutions, its ability to bypass the need for a DNA template, and its relatively high error rate of1.510-3SNPs/kb made it an excellent target for improving directed evolutionary methods.4, 5 Our team was fascinated by these and many other abilities of this replicase, such as being RNA-based. This facette, particular, was unique to mutagenesis methodologies, and we thought it to be quite advantageous. As RNA is universal to all living organisms, we hypothesized that by using an RNA-based mechanism we could, one day, achieve an organism-agnostic system. Upon letting the idea simmer in our team’s brains for several weeks, we thought we could take it a step further and make the system as easy as an RNA transfection. In creating a self-selective system, the need for manual selection could be hypothetically bypassed, reducing protein engineering largely down to an initial transfection of a target gene. Continuing the stream of ideas, the growing enthusiasm, and accruing desire to lessen the burdens of protein engineering, we became committed to creating the DiCE system.
Design
During the beginning stages of our team’s project, we branched out our resources and designed several wet-lab and technical projects simultaneously to allot ourselves options. In these early stages, an initial construct design for Qβ was used for proof-of-concept work on the functionality of the beta subunit of Qβ replicase within our host organism, Escherichia Coli. Upon finding initially promising results, our group convened and chose to keep Qβ. In order to prove our hypotheses, we required, however, to optimize our construct design and implementation strategy to appease our goals and allow for a concrete proof-of-concept of directed evolution.
- Initial construct design
In the very early stages of the DiCE leg of the project, the design of the initial vector plasmid was created. Our initial desire was to create a schema to test for the evolutionary capabilities of Qβ in vivo, on a target gene of an antibiotic selection marker. To do so, the intended construct desired extensive cloning. The steps taken to successfully clone the construct are outlined below:
Step 1: A pET28a+ vector was ordered from New England Biolabs (NEB), while our part inserts Qβ replicase and MDV were ordered from Integrated DNA Technologies (IDT).
Step 2: The pET28a+ vector was opened up between the T7 promoter and terminator region using a high-fidelity Q5 Polymerase. The linearized vector was then cloned using Gibson Assembly to have an MDV fragment insert.
Step 3: The cloned vector was then reopened as before in the middle of the MDV region to create MDV-1 and MDV-2 flanking regions, allowing for a gene(s) of choice to be replicated by Qβ by acting as recognition sites on the mRNA.6, 7 One of these genes being that which encodes Qβ replicase itself, which was ligated into the vector via Gibson Assembly.
Step 4: For a third time, Gibson Assembly cloning was used to insert a chloramphenicol resistance gene, whose DNA fragment was PCRed from an in-house construct supplied by our host student group, BIOME.

Step 5: Our construct was, after each cycle of cloning, grown and selected for on kanamycin plates. After the final round of cloning and antibiotic selection, picked colonies were grown in kanamycin, mini-prepped, and then sequence-verified at our local sequencing facility: PAN.
- Optimized Design
In order to refine the original design to better match our ultimate goals, the vector plasmid design was refined to account for concerns with the dynamics and mutagenesis kinetics of the mechanism. Early questions and critics of the original system raised by advisors, interviewees, and ourselves included: how are we to account for the mutagenesis of the Qβ protein itself (Qβ replicase was originally expressed in the mutagenized mRNA)? How might the exponential proliferation of protein account for resistance rather than an optimized protein? And how might the exponential kinetics of the system impact the longevity of the cells? We tackled these problems among others with creating the constructs outlined below:
Step 1: Using the original pRNA construct as the template plasmid vector, the high fidelity Q5 polymerase was utilized to linearize backbones for both Qβ and No Qβ constructs. The former’s backbone was PCRed with Gibson overhangs from the T7 promoter and terminator in opposing directions. The latter, was PCRed similarly, except with the addition to include the MDV-1 and MDV-2 regions.

Our 2019 iGEM team, with three high school interns front and center :) Step 2:The Qβ fragment was also PCRed with Gibson overhangs from the original pRNA construct to prepare for Gibson Assembly. Other genes, such as chloramphenicol and ampicillin resistance markers (CMR and AmpR respectively), intended to undergo mutagenesis for proof-of-concept experimentation underwent additional preparatory steps. Another caveat is that when PCR-ing these particular DNA fragments, the Gibson overhangs were designed such that the gene would be ligated into the vector backbone in a reverse complimentary orientation. This would allow us the ability to definitively prove Qβ’s replicative functionality through protein synthesis of antisense mRNA, which can not be created by typical RNA polymerases found in E. coli.
Step 3: Target genes would not only be utilized as is within the DiCE system but as a proof-of-concept for the mutagenic capabilities of Qβ, single nucleotide polymorphisms (SNPs) were introduced to the genes (e.g. CMR and AmpR) using the Quick Change Mutagenesis Kit.
Step 4: Fragments were then ligated into their respective backbones using Gibson Assembly.
Step 5: No Qβ as a plasmid is not pragmatically useful as the vector backbone is the same as that of Qβ’s. Rather, this construct serves to propagate a target gene in its original form for practical reasons. This gene within the MDV flanks is then selectively amplified in vitro, using a T7 High-Scribe Kit to prep for transfection and subsequent evolution. In other words, the mRNA itself, serves as the applied construct.

- Mechanism
Working towards our goal of developing a successful in vivo directed evolution system using Qβ replicase, we attempted to optimize initial constructs to create a streamlined process for set-up, mutagenesis, and selection. The envisioned process is depicted below:

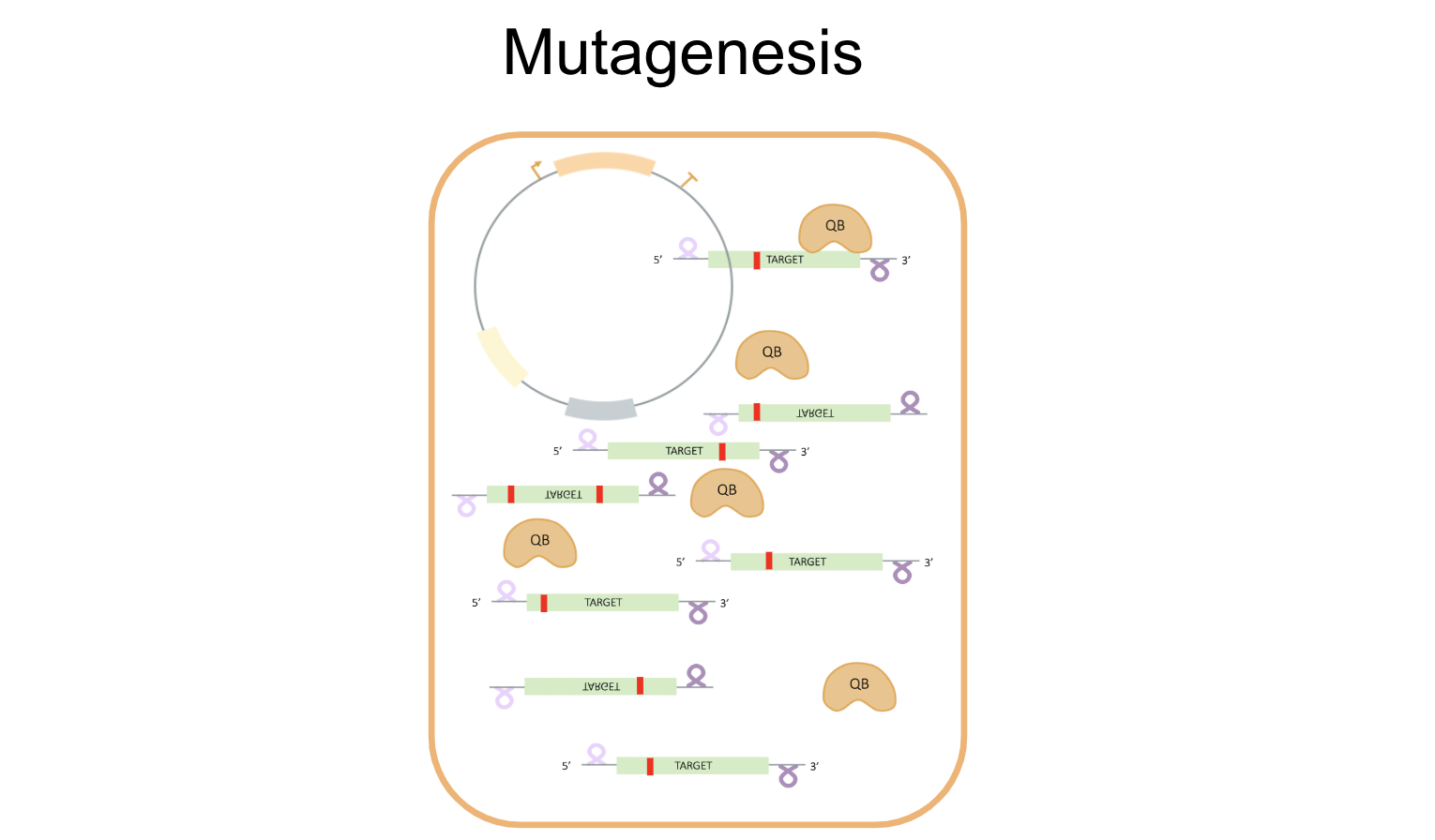


Experiment
Experiment 1: Does Qβ Self-Replicate in E. coli?
To summarize this first experiment, we tested for the ability of Qβ replicase to replicate sense mRNA, transcribed off a plasmid vector. In order to do so, we tested for the antisense strand of RNA, which could only form as a result of replication with Qβ. This was accomplished by, in short, synthesizing cDNA from extracted RNA after one 12-hour growth phase in a chloramphenicol dosage assay. Using primers, specific to either sense or antisense RNA strands to synthesize the cDNA, we could detect the presence of antisense RNA by running gels of both antisense and sense cDNA.
It is important to note that this experiment was run with the initial construct design, pRNA, as described in the design section above. For simplicity, the target gene is shown in green while the gene encoding for Qβ replicase is omitted as it is not necessary to mention for this experiment. The major steps taken for this experiment are depicted below.



Experiment 2: Can Qβ replicase Increase Antibiotic Resistance?
While it does not substantiate the hypothesized mechanism depicted in the design section, it does suggest to our team that perfecting the DiCE system is possible within E. coli. It is important to note, however, that this experiment was done using pRNA, and not the optimized constructs. A more comprehensive set of experiments with the new construct designs, No Qβ and Qβ have been designed and are awaiting the success of our mRNA gene delivery protocol for E. coli.
To summarize the experiment, several rounds of chloramphenicol dosage assays were done to provide an increasing selective pressure to increase resistance by optimizing the gene (again, this was using the pRNA construct and, therefore, can’t substantiate this hypothesis without sequencing data). After each round, cells that had survived in the highest concentration of chloramphenicol were moved into the next assay at even higher concentrations.
Experiment 3: Can mRNA Gene Delivery Be Accomplished in E. Coli and Maintained Qβ replicase?
This experiment is still being troubleshooted due to the overwhelming lack of literature on the subject of mRNA gene deliver in E. coli. Currently, this experiment is being attempted under several varying conditions and is also awaiting new reagents and parts. It is crucial to note that this experiment is utilizing the new construct designs, No Qβ and Qβ. Our protocol for creating electrocompetent cells can be found here in a Google doc as it is still under experimentation and not ready for publication on our website. Additionally, our electroporation protocol can be found here. Conditions tested are shown below:

Results
Currently, results have largely been impeded with the optimized constructs due to time and difficulty successfully establishing a working procedure of gene delivery into our electrocompetent E. coli. To the best of our knowledge, a single paper has been published within the last 40 years on the transfection of E. coli with mRNA by Akira Taketo, which has greatly limited the foundation from which we could work upon.8 This procedure is still under experimentation and being modified in order to move on to additionally planned experiments and proofs-of-concept in order to validate the hypothesized mechanism above.
Despite these current impediments to the in vivo portion of the DiCE project, data was gathered with the initial construct design. This data shows promise that Qβ replicase functions within bacteria and qualitatively suggests that mutant proteins were created for increased resistance to chloramphenicol. The latter, however, can’t be validated like the former due to a lack of sequencing data and inherent flaws of the initial construct design.
Experiment 1: Results

Experiment 2: Results:

Experiment 3: Results:
We are still gathering data and troubleshooting. We will edit this page when publishable data is available. :)