Team:Warwick/Results
Results
Synthetic Fatberg DNA Extraction
Our endeavour to degrade fatbergs using the power of synthetic biology began by contacting various wastewater treatment and management companies across the UK, with the hope of obtaining a fatberg sample. Eventually, we contacted United Utilities in Liverpool who offered to send us a sample. Whilst the fatberg was in transit we began to search for protocols concerning the extraction of DNA from samples with a high lipid content and were lucky enough to contact Qiagen who generously offered to sponsor us by sending a 'PowerFaecal pro DNA extraction tool kit'.
Following thorough advice concerning the difficulties surrounding DNA extraction from fatbergs from Dr. Justin Pachebat (University of Aberystwyth) and Dr. John Love (University of Exeter) we decided to construct a synthetic fatberg to test the proficiency of our Qiagen DNA extraction kit. After spiking 21 different synthetic fatberg aliquots with E. coli cells and extracting the DNA using the kit, we nano-dropped each aliquot to check the DNA concentration and ran a gel to ensure we had extracted DNA. Unfortunately, we were unable to isolate any DNA from the trial.
Fearing that the DNA samples were too low to be detected in our gels, we amplified the DNA using 16S RNA PCR and ran another gel on the products but were again, unsuccessful as shown by figure 1.

Did We Extract Actual Fatberg DNA?
Following the approval of our risk assessments to work on an actual fatberg sample in a biosafety level 2 laboratory, we undertook another round of DNA extractions using our Qiagen toolkit on our sample, although we had doubts due to the kit being unsuccessful during the trial run. Unfortunately, the DNA yields were low and as seen within figure 2, our extraction failed. This prompted us to contact Dr. Justin Pachebat once again, who revealed the protocol for fatberg DNA was surprisingly complex, requiring the use of liquid nitrogen, proteinase K based lysis buffers and multiple rounds of phenol chloroform based extractions, in addition to many ethanol precipitation steps. After signing an MTA (material transfer agreement) with him, Dr. Pachebat agreed to send us DNA his lab extracted from the Whitechapel fatberg.

Sequencing The Largest Fatberg In The World
We prepared a DNA library from the sample of fatberg DNA provided by Dr. Pachebat for sequencing, which was loaded onto a flow cell comprising a MinION device, generously donated by another of our sponsors - Oxford Nanopore. Following base-calling, we obtained 12Gb of sequences from the fatberg metagenome - that's approximately four times the length of the human genome! we identified 22 predicted lipase coding sequences, 21 of which are completely novel. This information was obtained by checking these sequences against the BLAST protein database in NCBI (National Center for Biotechnology Information). These sequences were revealed to have between 55 - 84% identity to the nearest hits. Several bacterial and yeast species were also observed within the metagenome, some of which were the same species our candidate lipases were derived from, following their identification from various literature sources.
After contacting Dr. Chris Quince based at the University of Warwick we were provided with a CPU computer cluster for metagenomic assembly. Following assembly, we utilised the software 'kraken2' to assign the reads and the contigs from the assembly to compile a phylogenetic tree of all the species found within our raw sequence reads. Several species were identified, including Pseudomonas fluorescens, which Thermostable lipase A (TliA) - a previous iGEM part (BBa_K258006) - is derived from.
Additionally, whilst we were waiting for both our fatberg sample from United Utilities and fatberg DNA provided by Dr. Pachebat, we searched various databases and literature sources for other candidate lipases to clone into our E. coli cells. Our starting point for this was the Thermostable lipase A (TliA) derived from the bacterial species Pseudomonas fluorescens - an iGEM part previously used by other teams such as Sheffield, Stuttgart and KAIST. BLAST searching TliA on the NCBI database revealed some similar lipases which were considered as potential candidates for our cloning strategy, including a lipase precursor from a compost metagenome. Other lipases and lipase-producing species were identified from research papers investigating industrial wastewater treatment plants, lipid-rich wastewater and restaurant wastewater. The complete list of selected lipases can be viewed on our design page
Did We Clone The Lipases Successfully?
Yes and no
We began our cloning strategy by obtaining a plasmid backbone from the Corre group, based at the University of Warwick. This backbone, named pJCC005, is used for cloning with both E. coli and Streptomyces cells. Consequently, we decided to design primers to amplify the part of the backbone we needed to create a new backbone optimised for transformation into our E. coli cells, removing all the Streptomyces-related genes. This process was harder and more time-consuming than anticipated due to the size of the pJCC005 backbone, requiring us to amplify the backbone in two parts and ligate them back together via Gibson assembly, as shown in figure 7. Despite the challenge we were able to successfully make our own, new backbone: pJC_BB12, which figure 8 depicts.

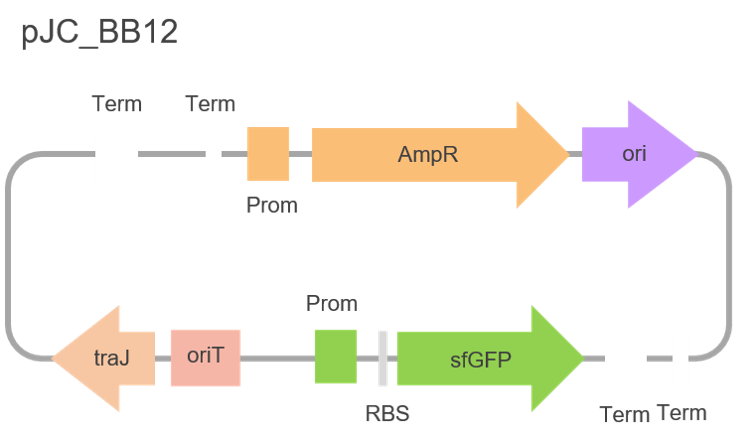
pJC_BB12 has a 'glpT' promoter for constitutive expression of our lipases and contains a gene encoding the super-folded green fluorescent protein (sfGFP), which conveniently made our bacterial colonies fluoresce green, facilitating our selection process of cells containing our lipases. We also hoped the fluorescent property of sfGFP would allow us to track both the expression and movement of our lipases.
We had two strategies for cloning our lipases into our backbone. Firstly, at the N-terminus of sfGFP with each of our lipases possessing a ribosome binding site (RBS), as well as a start and stop codon to produce two separate proteins - a lipase and sfGFP. We also wanted to fuse our lipases to the N-terminus of sfGFP to create one green fluorescent protein. In this scenario, both the lipase and sfGFP genes share an RBS, stop and start codons. In order to achieve this, we had to amplify our lipases in two slightly different ways for Gibson assembly. We decided to do this using both a Phusion polymerase - a high fidelity enzyme - and MyTaq polymerase - a more robust enzyme - and take the cleanest PCR products forward, as shown in the gel below.
We were really pleased to find we had successfully amplified seven of our eight lipases for both insertion next to the N-terminus of sfGFP and fusion at the N-terminus of sfGFP, shown in figure 9. However, we were a little baffled when we plated our transformed E. coli cells and incubated them only to discover that nothing grew on any of our plates. We repeated our transformation a few times and to our amazement the same result kept repeating itself. Additionally, after testing the dead cells on our tributyrin agar (see design) we were surprised to see that no lipase activity was found. This led us to hypothesize that perhaps the accumulation of these lipases inside our cells was toxic. Interestingly, figure 10 shows the successful cloning of a non-functional version of our compost metagenome lipase precursor into our backbone. This discovery was surprising, given we were able to successfully grow colonies of our transformed cells - a further suggestion that the accumulation of functional lipases within our cells was toxic.




A New Strategy
To combat this, we thought about using secretion tags to export the lipases from our cells and prevent their accumulation, as well as putting our lipases under the control of an inducible promoter. Consequently, our next strategy involved cloning a small selection of our candidate lipases (since at this point we were pressed for time) into a new vector: pET151/D-TOPO, as shown below. The lipases used included Lipase A, chain A from Candida antarctica (CALA), an alkaliphilic lipase from Bacillus subtilis (BSAL), our compost metagenome lipase precursor (CMLP) and the Thermostable lipase A (TliA) from Pseudomonas fluorescens (please see our design page for justifications on this selection of lipases). This new backbone included a T7 promoter, enabling the induction of lipase expression with Isopropyl-β-D-thiogalactoside (IPTG). We decided to use this backbone following discussions with Dr. Love from Exeter University, who advised we engineer our bacteria such that they secrete our lipases in a controlled manner. This would not be possible if the lipases were under the control of a constitutive promoter like 'glpT'.

Following the amplification of our selection of lipases to be cloned into the pET151/D-TOPO vector (figure 11), figure 12 reveals the products obtained following cloning, miniprepping of our clones and performing a PCR using primers specific to each of our chosen lipases. After obtaining Sanger sequencing data of each of our clones, we were really pleased to discover that we successfully cloned in three of our lipases, as well as a fragment of TliA. These results further supported the hypothesis that our bacterial cells were dying due to the constitutive expression and accumulation of our lipases. Our next step was to characterise the lipase activity of our engineered E. coli expression strains (BL21 star) using both a quantitative and qualitative assay of our own design.


Determining Lipase Function
Figure 13 depicts the observed lipase activity from our E. coli cells when induced with IPTG compared to our non-induced cells, more evidence to support the hypothesis that the accumulation of these lipases are indeed toxic.
Alongside this, to demonstrate and ensure our new parts containing our functional lipases worked, we developed a quantitative assay to determine the efficiency of each of our enzymes by measuring the kinetic parameters of Km, kcat and Vmax. This was achieved using the substrate p-nitrophenol octanoate, which contains an ester bond chemically identical to those found in lipids constituting fatbergs. Following hydrolysis by our lipases, p-nitrophenol octanoate is broken down into the products p-nitrophenol and octanoic acid. p-nitrophenol is yellow in solution, compared to p-nitrophenol octanoate, which is colourless. This property allowed us to develop a spectrophotometric assay to characterise the activity of our lipases by measuring the absorbance of p-nitrophenol at a wavelength of 400nm. The more p-nitrophenol octanoate is cleaved by our lipases, the more yellow our solution.
This led us to produce a standard absorption curve for TliA, a previous iGEM part. This spectrophotometric assay provided quantitative data required to characterise this lipase, having been previously used by Sheffield iGEM in 2014, as well as Stuttgart and KAIST iGEM. The parameters of Km, kcat and Vmax, determined from the Lineweaver-Burk plot shown in figure 15 have since been uploaded to the iGEM registry and are as follows: Km : 0.256mM, kcat : 0.097mM/min/mg and Vmax : 0.0049mM/min.
Following cell lysis, induction with IPTG and construction of the standard curve, shown in figure 14, a Lineweaver-Burk plot for BSAL (figure 16) was determined, from which the kinetic parameters of Km (0.349mM) and Vmax (0.0036mM/min) were determined. We were unable to determine kcat due to time constraints, which prevented us from purifying the enzyme and hence, calculating the amount of enzyme used in the assay, which was needed to calculate this parameter. No Lineweaver-Burk plot was constructued for CALA or CMLP and no kinetic parameters were determined. This was because the absorbance readings obtained for either enzyme were bizarre, showing inconsistent rates which most likely can be attributed to the fact that both lipases catalysed the reverse reaction generating p-nitrophenol octanoate, rather than p-nitrophenol and octanoic acid.




Testing The New Lipases
In addition to cloning our lipases into our E.coli expression strain BL21 star and characterising the activity and efficiency of our lipases using a spectrophotometric assay to derive the kinetic parameters of Km, Vmax and kcat we also wanted to assess whether our engineered bacteria could survive and better yet, grow in oil. After all, these bacteria will need to survive within a fatberg! With this in mind, we carried out a series of oil media experiments, measuring the population growth of E. coli at different oil concentrations using CFU (colony forming units) counts.
From here, we used the logistics equation to produce our model depicting the growth of bacteria at varying oil concentrations (please see our model page). This model was used to help us determine the optimal oil concentration to grow our engineered bacteria in. After consulting our model and talking to Dr. Kalesh Sasidharan, we decided to use a MicrobeMeter from Humane Technologies, to measure the growth of our engineered bacteria at an oil concentration of 0.5%. This concentration was chosen after consulting the model and identifying that as an optimal oil concentration. Additionally, we decided to use a Microbemeter to measure bacterial growth as Dr. Sasidharan advised that using CFU counts would be too labour intensive and time-consuming, in addition to us not being able to obtain a complete data-set needed to construct a complete growth curve.
Using the MicrobeMeter we first measured the growth of each of our engineered cells containing a non-induced construct, shown in figure 17 below. The results of this experiment revealed that the engineered bacteria containing CMLP and CALA did not follow a traditional growth curve, unlike BSAL, but rather showed 'peaks of activity' before their absorbance declined, signifying that the cells had died. Despite the constructs remaining uninduced, the presence of each construct severely affected growth rate.
We then decided to repeat the experiment and induce each of our cells, adding 10μl of 1mM IPTG five hours into growth, which can be observed in figure 18. This was done to determine the effect of lipase expression and accumulation within our cells. Interestingly, we were surprised to find that the growth curves for CALA and CMLP were very similar to the growth curves obtained for the same, uninduced cells. The growth curves for BSAL differed but the cells containing the BSAL construct died as expected upon induction with IPTG, shown by the decrease in absorbance. There was no significant difference between the doubling times obtained for both the induced and non-induced cells, as shown in the table below. The limited difference in growth rates between our induced and non-induced cells, coupled to the remarkable similarity between both the induced and non-induced CMLP and CALA growth curves led us to theorize that our constructs were being transcribed without the use of IPTG.


Do Lipases Limit The Growth Of Bacteria?
The bizarre growth curves led us to believe that perhaps the lipases were affecting a process present in only a single stage of bacterial growth. To investigate the effect of our lipases within our engineered cells during the lag phase (the initial phase of growth) we decided to inoculate 10ml of LB containing ampicillin with each of our constructs and immediately induce our cells with 10μl of 1mM IPTG. This experiment yielded interesting results, revealing the cells still grew, unaffected by induction, as shown by an increase in turbidity. These results led us to hypothesize that a process during the lag phase was resulting in the death of our engineered cells upon the production of our lipases.
What was also suspicious was the seemingly perfect coordination of death or stagnation between our engineered bacterial cells containing either CALA, CMLP or BSAL. This is highlighted in the growth curves of the uninduced cells containing either CALA or CMLP constructs. Both engineered cells reached the same optical density at approximately 0.15, followed by a decrease in absorbance. Consequently, we hypothesized that our lipases were being induced by a process triggered by quorum sensing. Unfortunately, we were unable to further test this theory due to time constraints.
Summary
Consequently, from sequencing fatberg DNA to identifying and successfully cloning candidate lipases, we have obtained valuable data strengthening the potential of our engineered bacteria to degrade fatbergs. We have managed to characterise the presence and activity of each of our lipase constructs, obtaining both qualitative and quantitative data from a tributyrin assay and spectrophotometric assay using p-nitrophenol octanoate. Additionally, we characterised the previous iGEM part TliA, created new, validated constructs, and demonstrated the ability of our engineered bacteria to degrade lipid molecules via the hydrolysis of ester bonds. Future development of our project may include adding a secretion tag to our lipase sequences in order to prevent their accumulation inside bacterial cells, avoiding cell death.
We have laid the foundations for tackling the fatberg crisis using the power of synthetic biology and we hope our research can be implemented and carried forward to benefit both the local and global community in the future. However, from both our research, model and public outreach activities we have understood that degrading fatbergs using lipases only provides a short term solution to these greasy nightmares. A more successful, long term strategy would be a focus on fatberg prevention schemes through education of the general public, which we have established via the continuous implementation of our human practices.
Contact Us



Sponsors













