


Inspiration
One day, our team leader heard an article about Manila hemp. As a member of the banana family, Manila hemp fibers have many excellent characteristics. So he thought if Manila hemp belongs to banana family, whether the fiber of banana stalk has the same properties and can extract high-quality fiber.
Fortunately, one of our team members is from Guangdong Province. His hometown is a banana producing area. Almost every family has planted a large number of bananas. Once he heard that the whole body of the banana planted is treasure. The banana leaf can also be used to deal with the wound, can be used to wrap traditional Chinese rice-pudding. Banana root is a kind of medicinal material. It can clear away heat and detoxify. Ancient Chinese medicine is also used to treat calculus. But only the stalk of banana, it can make some coarse linen cloth clothes before. Now, with the fiber of chemical industry, no one will choose such clothes. And every year, a large number of banana stalks are cut down, which can only be used as fertilizer in the field for the next year.
After the he told us about this problem, we looked up relevant information and found that 410 million people in the world eat bananas as staple food, and the annual stalk output reached 8.8 billion tons.

We also consulted our college's teachers who are studying ramie degumming technology. They also think that biological treatment of banana stalk to extract fiber is a problem worthy of study. And their ramie and our banana stalk have some similarities. We can do our research based on their relatively mature technology.
So we decided to use biological fermentation to make banana stalk into fine fiber, which can become people's beautiful clothes again in the future.
Fortunately, one of our team members is from Guangdong Province. His hometown is a banana producing area. Almost every family has planted a large number of bananas. Once he heard that the whole body of the banana planted is treasure. The banana leaf can also be used to deal with the wound, can be used to wrap traditional Chinese rice-pudding. Banana root is a kind of medicinal material. It can clear away heat and detoxify. Ancient Chinese medicine is also used to treat calculus. But only the stalk of banana, it can make some coarse linen cloth clothes before. Now, with the fiber of chemical industry, no one will choose such clothes. And every year, a large number of banana stalks are cut down, which can only be used as fertilizer in the field for the next year.
After the he told us about this problem, we looked up relevant information and found that 410 million people in the world eat bananas as staple food, and the annual stalk output reached 8.8 billion tons.
We also consulted our college's teachers who are studying ramie degumming technology. They also think that biological treatment of banana stalk to extract fiber is a problem worthy of study. And their ramie and our banana stalk have some similarities. We can do our research based on their relatively mature technology.
So we decided to use biological fermentation to make banana stalk into fine fiber, which can become people's beautiful clothes again in the future.
Description
Banana, a fruit with magical and hidden treasure.
According to Food and Agriculture Organization of the United Nations (FAO), the world's largest producers of bananas in 2017 were India and China, which together accounted for approximately 38% of total production.
In southern China, there grows massive banana trees, and every year after harvesting the fruit, the false stem of the banana tree will be cut off and abandoned on the farm, becoming a kind of agricultural waste.
By searching for the relative information on the internet and communicating with the banana planters, we aware that banana stems are not only huge in scale but also a treasurable raw material resource waiting to be treated and utilized.
The stalk contains abundant industrial precursor and crude materials, including cellulose, lignin, saccharides, proteins, and trace element.

Figure1&2: Banana are staple food for many people[1]
In southern China, there grows massive banana trees, and every year after harvesting the fruit, the false stem of the banana tree will be cut off and abandoned on the farm, becoming a kind of agricultural waste.
By searching for the relative information on the internet and communicating with the banana planters, we aware that banana stems are not only huge in scale but also a treasurable raw material resource waiting to be treated and utilized.
The stalk contains abundant industrial precursor and crude materials, including cellulose, lignin, saccharides, proteins, and trace element.
Figure1&2: Banana are staple food for many people[1]
Bananas’ castoff application[2]
Textile
Textile
Banana fiber is widely found in banana stalk, banana leaves and fruit shafts, which are natural cellulose fibers, and banana fibers have the characteristics of natural hemp fibers. It is a new environmental-friendly material for waste utilization.

Figure3&4: People use banana fiber to weave clothes[3][4]
Figure3&4: People use banana fiber to weave clothes[3][4]
Papermaking
The quality of banana fiber pulp is relatively stable. With the improvement of technology, its mechanical strengths are close to bamboo pulp, and banana stalk can be recycled as a new papermaking material. What's more, the emissions on its production line are environmentally friendly.

Figure 5&6: Banana fiber are used in papermaking industry[5]
Figure 5&6: Banana fiber are used in papermaking industry[5]
Fertilizer
The banana stalk waste residue and the microorganisms are put together to produce organic fertilizer, which increases the soil organic matter and improves the soil condition. Achieve the soil circulation of the trace elements.

Figure7&8: Stalk waste can be used as fertilizer[5]
Figure7&8: Stalk waste can be used as fertilizer[5]
Feed
Banana pseudo stems are rich in nutrients, and protein feed using banana stalks as the main raw material can meet the growth needs of feeding animals.

Figure9&10: Banana relative materials can feed animals[5][6]
Figure9&10: Banana relative materials can feed animals[5][6]
Energy
It is rich in a large amount of sugars that can be used to ferment ethanol or biodiesel, which is a green energy source.

Figure11&12: Derivations like ethanol are green energy[1]
Figure11&12: Derivations like ethanol are green energy[1]
Comprehensive Utilization
Lignin is separated and degraded to produce benzene-containing substances, which have great application prospects.
According to all the information we have collected, we think it’s necessary and meaningful to treat banana stem. Nowadays most of the banana stems are treated by chemical, such as strong acid and alkali. Chemical-treating not only produce a lot of pollution, but also is low in profit, and the products are poor quality. So we decide to design a green and environmental friendly biological method to process banana stem.
What do we want to get from recycling the bananas’ castoff, especially their stalk?
We focus on cellulose, a kind of polysaccharide which is widely found in plants, as our project goal. Cellulose is the most abundant and renewable natural organic polymer in nature. Besides plants, we can find it in marine organisms and microorganisms either. The chemical structure of cellulose is a large molecule, which is formed by glucose units with beta-1, 4-glycoside bond connecting them. In nature, plant cell wall is the main source of cellulose. [7]
We searched the related reference to look for the information about the cellulose of banana’s stalk, and we found that the content of cellulose in banana is close to ramie, which used widely in textile, and we inferred that the cellulose in banana can be utilized. Hence, we decided to extract the cellulose of the banana. But there are other problems. Compared with ramie, the banana has more lignin (A material mainly located between cellulose fibers as an anti-compression agent, consist of three kinds of phenylpropane units), so we had to remove it. Another content in stalk—pectin (a group of poly-galacturonic acids)—also need to be removed. Therefore, we designed a system which help us to handle this problem.
We searched the related reference to look for the information about the cellulose of banana’s stalk, and we found that the content of cellulose in banana is close to ramie, which used widely in textile, and we inferred that the cellulose in banana can be utilized. Hence, we decided to extract the cellulose of the banana. But there are other problems. Compared with ramie, the banana has more lignin (A material mainly located between cellulose fibers as an anti-compression agent, consist of three kinds of phenylpropane units), so we had to remove it. Another content in stalk—pectin (a group of poly-galacturonic acids)—also need to be removed. Therefore, we designed a system which help us to handle this problem.
How we design the system?
First, in order to remove the lignin and pectin in banana stalk, we need special enzyme to decomposed them. By searching lots of reference, we chose small laccase (hereinafter called SLAC), versatile peroxidase (VP) , and pectate lyase (one kind of pectate lyase called PelA). Our goal is to transferred these three genes which is corresponding with the three kinds of enzymes into our model microorganism, the Pichia pastoris.
So, we designed a secrete regulatory system, which use a kinds of signal peptide to make it, and we chose the unique signal peptide corresponding to different enzymes from the six signal peptides we prepared in advance. And in order to regulate the pH in fermentation process, we created a pH responding regulation system, by using some interesting pH-induced promoters. This part we will show elaborately in our design part.
So, we designed a secrete regulatory system, which use a kinds of signal peptide to make it, and we chose the unique signal peptide corresponding to different enzymes from the six signal peptides we prepared in advance. And in order to regulate the pH in fermentation process, we created a pH responding regulation system, by using some interesting pH-induced promoters. This part we will show elaborately in our design part.
Reference:
[1] https://image.baidu.com
[2] Recent Achievements and Analysis of Comprehensive Utilization of Banana Stalk Wastes, Chinese Journal of Tropical Agriculture, 2013, 33(7):63-67
[3] http://i-want.tw/project/view/id/1466
[4] https://www.walkerland.com.tw/article/view/68039?page=
[5] https://unsplash.com
[6] http://news.cctv.com/world/20070313/106440.shtml
[7] FU Shiyu Progress in Cellulose Research State Key Lab of Pulp and Paper Engineering,South China University of Technology,Guangzhou,Guangdong Province,510640
[2] Recent Achievements and Analysis of Comprehensive Utilization of Banana Stalk Wastes, Chinese Journal of Tropical Agriculture, 2013, 33(7):63-67
[3] http://i-want.tw/project/view/id/1466
[4] https://www.walkerland.com.tw/article/view/68039?page=
[5] https://unsplash.com
[6] http://news.cctv.com/world/20070313/106440.shtml
[7] FU Shiyu Progress in Cellulose Research State Key Lab of Pulp and Paper Engineering,South China University of Technology,Guangzhou,Guangdong Province,510640
Design
Genes and Chassis
As we mentioned before, to focus on cellulose and get high quality banana fiber, the most important work is removing lignin and pectin. But what kind of genes should we choose? After consulting papers, we choose pectin lyase A (pelA), small laccase (slac) and versatile peroxide (VP) to achieve our goal.
pelA: Pectin lyase A was found in Aspergillus nigra EIM-6. It can break ester bonds in pectin. We use it to deal with the pectin which cover the surface of the fiber. And it will generate the galacturonic acids.
The lyase acts on the fifth position β carbon atom of galacturonic acid by β racemization, so that the H on the β carbon atom is transferred to the oxygen atom of the glycosidic bond, and the glycosidic bond is broken, forming a double bond between C4 and C5 of galacturonic acid. It produces galacturonic acid oligosaccharide.
VP: Versatile peroxidase was found in Pleurotus ostreatus. Versatile peroxidase is a kind of peroxidase comprising activities of manganese and lignin peroxidases. It has interaction sites of those two. They have different mechanisms to decompose the lignin, but they oxidize it first. The oxidized lignin generates unstable free radicals and then undergo a series of non-enzymatic spontaneous cracking reactions, leading to oxidation and fracture of lignin polymers.

SLAC: Small laccase catalyzes oxidation of most model compounds of lignin with free phenols. The catalytic reaction contains four consecutive one-electron oxidations, oxidizing the phenol ring to generate the phenoxy group, an intermediate from the radical. Phenoxy group is a cationic group with oxygen as the center. It can usually undergo non-enzyme-catalyzed cleavage reaction, which eventually leads to the bond breaking between aryl group and α carbon atom.
We also need an appropriate expression vector. After discussion, we chose Pichia pastoris to be our chassis because of its clear and simple mechanism and strong ability to express enzyme. Pichia pastoris has been used in synthetic biology field for a long time because of its expression advantages. This time, we chose pichia pastoris because exogenous protein can be expressed in pichia pastoris with high quality and sizable quantity.

pelA: Pectin lyase A was found in Aspergillus nigra EIM-6. It can break ester bonds in pectin. We use it to deal with the pectin which cover the surface of the fiber. And it will generate the galacturonic acids.
The lyase acts on the fifth position β carbon atom of galacturonic acid by β racemization, so that the H on the β carbon atom is transferred to the oxygen atom of the glycosidic bond, and the glycosidic bond is broken, forming a double bond between C4 and C5 of galacturonic acid. It produces galacturonic acid oligosaccharide.
VP: Versatile peroxidase was found in Pleurotus ostreatus. Versatile peroxidase is a kind of peroxidase comprising activities of manganese and lignin peroxidases. It has interaction sites of those two. They have different mechanisms to decompose the lignin, but they oxidize it first. The oxidized lignin generates unstable free radicals and then undergo a series of non-enzymatic spontaneous cracking reactions, leading to oxidation and fracture of lignin polymers.
SLAC: Small laccase catalyzes oxidation of most model compounds of lignin with free phenols. The catalytic reaction contains four consecutive one-electron oxidations, oxidizing the phenol ring to generate the phenoxy group, an intermediate from the radical. Phenoxy group is a cationic group with oxygen as the center. It can usually undergo non-enzyme-catalyzed cleavage reaction, which eventually leads to the bond breaking between aryl group and α carbon atom.
We also need an appropriate expression vector. After discussion, we chose Pichia pastoris to be our chassis because of its clear and simple mechanism and strong ability to express enzyme. Pichia pastoris has been used in synthetic biology field for a long time because of its expression advantages. This time, we chose pichia pastoris because exogenous protein can be expressed in pichia pastoris with high quality and sizable quantity.
Parts Design
In our design, to get these enzymes secreted out of cells to decompose pectin and lignin, we need a signal peptide to combine with each enzyme to achieve the transmembrane transportation of enzyme. That’s why we insert a piece of signal peptide sequence.
And to control whether parts work or not, AOX1 promoter, a methanol inducible promoter, was used. We also add a His-Tag in its tail. His-Tag can add several special amino acids that chelate metal ions and make the proteins easier to be attached to chromatography media. In other words, it will be easier to purify and collect the proteins.
In order to develop an efficient Bananas’ castoff-process system, we want Pichia pastoris to secrete more enzymes, so we find six different signal peptides from literatures, link each signal peptide sequence to each enzyme gene and get eighteen combinations. In this step, we transfer every combination into expression vector respectively, and add methanol to induce the expression, and measure the enzyme activity. In this way, we can know which combination can produce the enzymes with highest activity.

And to control whether parts work or not, AOX1 promoter, a methanol inducible promoter, was used. We also add a His-Tag in its tail. His-Tag can add several special amino acids that chelate metal ions and make the proteins easier to be attached to chromatography media. In other words, it will be easier to purify and collect the proteins.
In order to develop an efficient Bananas’ castoff-process system, we want Pichia pastoris to secrete more enzymes, so we find six different signal peptides from literatures, link each signal peptide sequence to each enzyme gene and get eighteen combinations. In this step, we transfer every combination into expression vector respectively, and add methanol to induce the expression, and measure the enzyme activity. In this way, we can know which combination can produce the enzymes with highest activity.
System Development
We have designed our parts and determined enzyme activity, but just linking them together is not enough. The problem lies that the product of pectin degradation is galacturonic acid, which will decline pH. In that case, what we need is a pH feedback regulation system, which will enable us to regulate pH. We will explain how we design this system and how it works as follow.
We choose pGAP promoter (This promotor does not need to induce expression) instead of AOX1 as the promoter of pelA gene considering of the usage in industry, because adding extra material into fermenter tank regularly means higher financial and labor cost. PgpdA promoter (a promoter works well when pH lower than 5) replaces AOX1 as the promoter of VP gene and SLAC gene. There is a new alkali gene with pgas promoter, which will express when pH is near 2.
Our design is about one device including four parts.

For starter, we chose the promoter pGAP to initialize the whole system. The promotor can initialize the expression of the first enzyme, pela, pectin lyase. It can hydrolyze the pectin into galacturonic acid, which is acidic. The pH value will get lower and lower.
When it reaches 5, the promoter PgpdA will be activated, and initialize the expression of versatile peroxidase and small laccase. Both of them are oxidases that can oxidize the lignin, and the combination will enhance the efficiency. Since there are many ether bonds in lignin, its oxidation will generate a lot of carboxyl groups and continue getting PH value decrease.
We cannot let pH get too low for Pichia pastoris to survive, so we’re going to need a regulator to increase the pH value again. That’s why we add the fourth part. Once the pH value reaches 2, then this promoter Pgas will be activated and highly expressed an alkali protein. In a word, if the system works, it will repeat this circulation until all the lignin and pectin are oxidized or decomposed.
To show whether the promotor works,we add green fluorescent protein instead of pelA, VP and SLAC after the promoters as a reporter.

We choose pGAP promoter (This promotor does not need to induce expression) instead of AOX1 as the promoter of pelA gene considering of the usage in industry, because adding extra material into fermenter tank regularly means higher financial and labor cost. PgpdA promoter (a promoter works well when pH lower than 5) replaces AOX1 as the promoter of VP gene and SLAC gene. There is a new alkali gene with pgas promoter, which will express when pH is near 2.
Our design is about one device including four parts.
For starter, we chose the promoter pGAP to initialize the whole system. The promotor can initialize the expression of the first enzyme, pela, pectin lyase. It can hydrolyze the pectin into galacturonic acid, which is acidic. The pH value will get lower and lower.
When it reaches 5, the promoter PgpdA will be activated, and initialize the expression of versatile peroxidase and small laccase. Both of them are oxidases that can oxidize the lignin, and the combination will enhance the efficiency. Since there are many ether bonds in lignin, its oxidation will generate a lot of carboxyl groups and continue getting PH value decrease.
We cannot let pH get too low for Pichia pastoris to survive, so we’re going to need a regulator to increase the pH value again. That’s why we add the fourth part. Once the pH value reaches 2, then this promoter Pgas will be activated and highly expressed an alkali protein. In a word, if the system works, it will repeat this circulation until all the lignin and pectin are oxidized or decomposed.
To show whether the promotor works,we add green fluorescent protein instead of pelA, VP and SLAC after the promoters as a reporter.
MODEL
To find the most suitable expression level instead of the highest, we build a model to stimulate the whole process and tend to find the match that saves as more substrates as possible. If you want to learn more, please move to the MODEL page.
Fermentation
Fermentation is an inevitable part in real industry. So we simulated the fermentation in the laboratory, by reducing its scale. First of all, we use physical way to roll the fiber, making it more loose. Then we boiled it at 120 Centigrade(240 Fahrenheit) for 20 minutes, keeping it at a high temperature and moisture circumstances. To let the fiber dry thoroughly, we put it into the oven and dry it for 60 Centigrade(140 Fahrenheit). We set different numbers of fiber, concentration of Pichia pastoris, adding 2 milliliter each day for 22 days. After fermentation, we compared the weight of before and after, finding out the best condition of fermentation.
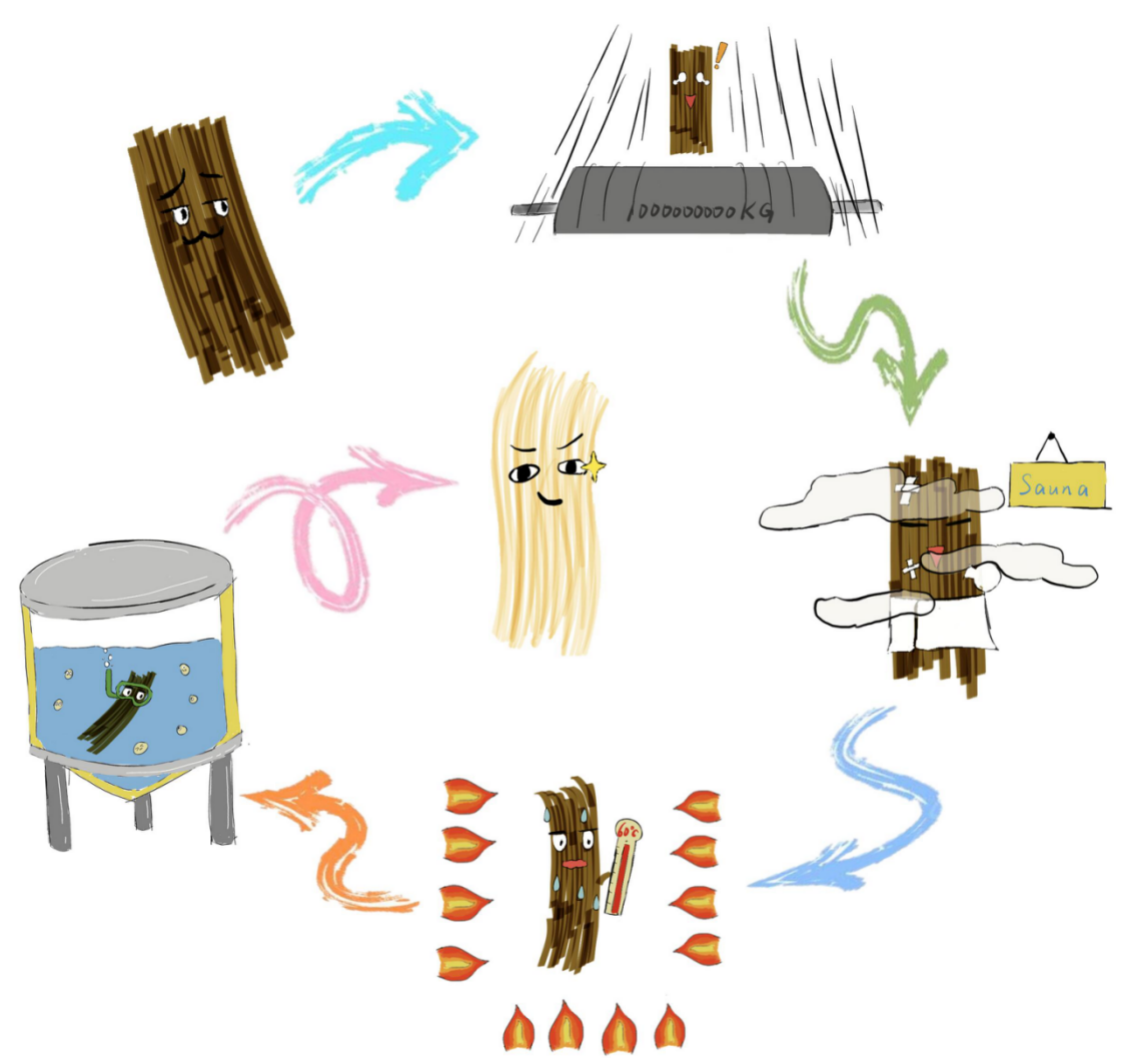
Reference: [1] Pei Fan. Research and application of in-situ biodegumming technology for ramie fiber[D].Hubei Huazhong University of Science and Technology, 2015. DOI:10.7666/d.D731116.
Reference: [1] Pei Fan. Research and application of in-situ biodegumming technology for ramie fiber[D].Hubei Huazhong University of Science and Technology, 2015. DOI:10.7666/d.D731116.
Experiment
Degradation system
The function of degradation system is degrade the lignin and pectin in banana stalk.
①Amplify
VP,SLAC,pelA
②Plasmid construction
Coding sequences: FLO10-SLAC, PHO5-SLAC, SUC2-SLAC, SLAC, FLO10-αpro-SLAC, PHO5-αpro-SLAC and same signal peptide with VP and pela—were cloned into the expression vector PPIC9K by Gene recombination, and then we transform the recombined PPIC9K plasmid into Pichia pastoris.

③Detection
We use spectrophotometric method to detect the enzyme activity of SLAC and VP. Also, we use ultrasonic crusher to break structure of cell and run SDS protein glue to detect the expression of proteins. As for pelA, we use DNS titration method to detect the enzyme activity of pelA.
Culture:First of all, we transfer the Pichia we want to BMGY medium. After expanding the culture, we transfer the Pichia with different gene into specific BMM medium.
According to the expression phenotype of the small laccase, we add copper ion to enhance its expression. Besides, we add few ABTS into the medium, so that we can visualize whether we induce active small laccase.
As for versatile peroxidase, we add manganese ion and heme chloride to help its expression.
Culture:First of all, we transfer the Pichia we want to BMGY medium. After expanding the culture, we transfer the Pichia with different gene into specific BMM medium.
According to the expression phenotype of the small laccase, we add copper ion to enhance its expression. Besides, we add few ABTS into the medium, so that we can visualize whether we induce active small laccase.
As for versatile peroxidase, we add manganese ion and heme chloride to help its expression.
PH responding system
This system makes the engineering bacteria work automatically.
①promoter selection
After searching numbers of paper, we selected pGAP promoter, PgpdA promoter and Pgas promoter to achieve this goal. The pGAP promoter had the highest expression in pH=7, the PgpdA promoter had the highest expression in pH=5, and the Pgas promoter had the highest expression in pH=2. In addition, because the content of pectin in banana stem is less than lignin, and located in the outside of banana fiber, we decided to decompose pectin first, and then decompose lignin to ensure the maximum decomposition efficiency. The pGAP promoter was linked to the pelA gene, the PgpdA promoter was connected to the VP and SLAC genes, and the Pgas promoter was linked to the alkali production gene. First of all, in the case of pH=7, pGAP initiated the expression of pelA, secreted Pectinase to decompose pectin, and pectin was decomposed into galacturonic acid. Galacturonic acid can reduce the pH of the environment. When pH decreased to 5, the expression of PgpdA promoter was initiated and SLAC and VP were secreted to decompose lignin in banana stem. If the pH value decreased too low, at 2, the Pgas as an insurance device began to start, resulting in a large number of alkali-producing gene expression, pH returned to 5, PgpdA normal operation. Due to the recovery of pH, the expression of pelA was lower than that of SLAC and VP.
②Construction of whole path
We finally put Pgap-Kozak-FLO10-αpro-pelA-FLO10-SLAC-TT-Pgas-Alkali-TT(Ⅰ)and Pgap-Kozak-PHO1-pelA-FLO10-SLAC-TT-Pgas-Alkali-TT(Ⅱ) into expression plasmid pGAPZαA, and transform the vector into Pichia pastoris to detect the effect of our project.

①promoter selection
After searching numbers of paper, we selected pGAP promoter, PgpdA promoter and Pgas promoter to achieve this goal. The pGAP promoter had the highest expression in pH=7, the PgpdA promoter had the highest expression in pH=5, and the Pgas promoter had the highest expression in pH=2. In addition, because the content of pectin in banana stem is less than lignin, and located in the outside of banana fiber, we decided to decompose pectin first, and then decompose lignin to ensure the maximum decomposition efficiency. The pGAP promoter was linked to the pelA gene, the PgpdA promoter was connected to the VP and SLAC genes, and the Pgas promoter was linked to the alkali production gene. First of all, in the case of pH=7, pGAP initiated the expression of pelA, secreted Pectinase to decompose pectin, and pectin was decomposed into galacturonic acid. Galacturonic acid can reduce the pH of the environment. When pH decreased to 5, the expression of PgpdA promoter was initiated and SLAC and VP were secreted to decompose lignin in banana stem. If the pH value decreased too low, at 2, the Pgas as an insurance device began to start, resulting in a large number of alkali-producing gene expression, pH returned to 5, PgpdA normal operation. Due to the recovery of pH, the expression of pelA was lower than that of SLAC and VP.
②Construction of whole path
We finally put Pgap-Kozak-FLO10-αpro-pelA-FLO10-SLAC-TT-Pgas-Alkali-TT(Ⅰ)and Pgap-Kozak-PHO1-pelA-FLO10-SLAC-TT-Pgas-Alkali-TT(Ⅱ) into expression plasmid pGAPZαA, and transform the vector into Pichia pastoris to detect the effect of our project.
Fermentation
We put banana stalks into fermentation device and observe the changes compared with control group.
①fermentation preparation
Pretreatment of banana stalk:
As for versatile peroxidase, we add manganese ion and heme chloride to help its expression.
1. Banana stalk was pretreated by mechanical rolling.
2. Put the stalk that has been mechanically rolled into the autoclave for high temperature and damp heat treatment, 121 ℃, 2 hours.
3. In an oven at 60 ℃ and dry for 12 hours.
4.Take out and weigh.
Engineering bacteria preparation:
1. The yeast which had been successfully transformed by electricity in the seed preservation tube was put into the bmgy medium and cultured for 12 hours.
2. Transfer yeast from bmgy to BMM medium.
②banana stalk fermentation
1. Put the pretreated banana stalk into the BMM medium which has been inoculated with yeast, and put it into the constant temperature shaker.
2. Every 24 hours, add 1 / 100 of methanol.
3. After about 12 days, take out the processed banana stalk, put it into an oven at 60 ℃, and dry for 72 hours.
4. Take out and weigh, and determine the components.
①fermentation preparation
Pretreatment of banana stalk:
As for versatile peroxidase, we add manganese ion and heme chloride to help its expression.
1. Banana stalk was pretreated by mechanical rolling.
2. Put the stalk that has been mechanically rolled into the autoclave for high temperature and damp heat treatment, 121 ℃, 2 hours.
3. In an oven at 60 ℃ and dry for 12 hours.
4.Take out and weigh.
Engineering bacteria preparation:
1. The yeast which had been successfully transformed by electricity in the seed preservation tube was put into the bmgy medium and cultured for 12 hours.
2. Transfer yeast from bmgy to BMM medium.
②banana stalk fermentation
1. Put the pretreated banana stalk into the BMM medium which has been inoculated with yeast, and put it into the constant temperature shaker.
2. Every 24 hours, add 1 / 100 of methanol.
3. After about 12 days, take out the processed banana stalk, put it into an oven at 60 ℃, and dry for 72 hours.
4. Take out and weigh, and determine the components.
Protocol
1. Medium
a. LB (Luria-Bertani) medium

b. 10×YNB medium
134g YNB dissolve in 800 mL double distilled water, heated, stirred and dissolved, fixed volume to 1 L, filtered and sterilized and stored at 4 ℃.

c. MD medium
15g Agar add to 900mL double distilled water, steam sterilized at 121℃ for 20 minutes. Cool to 60℃, and add 100mL 10×YNB, 2mL 500×B, 40mL 50% glucose, stored at 4 ℃.

d. YPD medium
Yeast extract 10g, peptone 20g, added to 900mL double distilled water, steam sterilized for 20 minutes. Add glucose to the final concentration of 20g. The solid medium is supplemented with Agar with a final concentration of 20g.

e. BMGY medium
Dissolve 10g glycerol, 1g yeast extract, 20g peptone, 100mL 1 mol/L phosphate buffer (pH 6.0) in 800mL double distilled water. Steam sterilized for 20 minutes, at 121℃. After cooling to 60℃, add 100mL 10×YNB, 2mL 500×B and store at 4℃.

f. BMMY medium
Dissolve 10g yeast extract, 20g peptone, 100mL 1mol/L phosphate buffer (pH 6.0) in 750 mL double distilled water. Steam sterilized for 20 minutes at 121℃. After Cooling to 60 ℃, add 100mL 10×YNB, 2mL 500×B, 50mL 10×M and store at 4 ℃.
 g. BMM medium
g. BMM medium
Dissolve 100mL 1mol/L phosphate buffer (pH 6.0) in 700 mL double distilled water. Steam sterilized for 20 minutes at 121℃. After Cooling to 60 ℃, add 100mL 10×YNB, 2mL 500×B, 50mL 10×M and store at 4 ℃.

b. 10×YNB medium
134g YNB dissolve in 800 mL double distilled water, heated, stirred and dissolved, fixed volume to 1 L, filtered and sterilized and stored at 4 ℃.
c. MD medium
15g Agar add to 900mL double distilled water, steam sterilized at 121℃ for 20 minutes. Cool to 60℃, and add 100mL 10×YNB, 2mL 500×B, 40mL 50% glucose, stored at 4 ℃.
d. YPD medium
Yeast extract 10g, peptone 20g, added to 900mL double distilled water, steam sterilized for 20 minutes. Add glucose to the final concentration of 20g. The solid medium is supplemented with Agar with a final concentration of 20g.
e. BMGY medium
Dissolve 10g glycerol, 1g yeast extract, 20g peptone, 100mL 1 mol/L phosphate buffer (pH 6.0) in 800mL double distilled water. Steam sterilized for 20 minutes, at 121℃. After cooling to 60℃, add 100mL 10×YNB, 2mL 500×B and store at 4℃.
f. BMMY medium
Dissolve 10g yeast extract, 20g peptone, 100mL 1mol/L phosphate buffer (pH 6.0) in 750 mL double distilled water. Steam sterilized for 20 minutes at 121℃. After Cooling to 60 ℃, add 100mL 10×YNB, 2mL 500×B, 50mL 10×M and store at 4 ℃.
g. BMM medium
Dissolve 100mL 1mol/L phosphate buffer (pH 6.0) in 700 mL double distilled water. Steam sterilized for 20 minutes at 121℃. After Cooling to 60 ℃, add 100mL 10×YNB, 2mL 500×B, 50mL 10×M and store at 4 ℃.
2. plasmid DNA extraction
a. Centrifuge at 10000×g for 1 minute at room temperature.
b. Decant or aspirate and discard the culture media.
c. Add 250μL solution I/RNase A. Vortex or pipet up and down to mix thoroughly.
d. Transfer suspension into a new 1.5mL micro centrifuge tube.
e. Add 250μL Solution II. Invert and gently rotate the tube several times to obtain a clear lysate. A 2-3 minute incubation may be necessary.
f. Add 350μL Solution III. Immediately invert several times until a flocculent white precipitate forms.
g. Centrifuge at max speed for 10 minutes. A compact white pellet will form. Promptly proceed to the next step.
h. Insert a DNA Mini Column into a 2mL Collection tube.
i. Transfer the cleared supernatant from Step 7 by carefully aspirating it into the DNA Mini Column.
j. Centrifuge at max speed for 1 minute.
k. Discard the filtrate and reuse the collection tube.
l. Add 500μL HBC Buffer.
m. Centrifuge at max speed for 1 minute.
n. Discard the filtrate and reuse the collection tube.
o. Add 700μL DNA Wash Buffer.
p. Centrifuge at max speed for 1 minute.
q. Discard the filtrate and reuse the collection tube.
r. Repeat steps 15-17 for a second wash.
s. Centrifuge the empty DNA Mini Column matrix before elution. Residual ethanol may interfere with downstream applications.
t. Transfer the DNA Mini Column to a clean 1.5mL microcentrifuge tube.
u. Add 30-100μL sterile deionized water directly to the center of the column membrane.
v. Let sit at room temperature for 1 minute.
w. Centrifuge at max speed for 1 minute.
x. Store at -20℃
b. Decant or aspirate and discard the culture media.
c. Add 250μL solution I/RNase A. Vortex or pipet up and down to mix thoroughly.
d. Transfer suspension into a new 1.5mL micro centrifuge tube.
e. Add 250μL Solution II. Invert and gently rotate the tube several times to obtain a clear lysate. A 2-3 minute incubation may be necessary.
f. Add 350μL Solution III. Immediately invert several times until a flocculent white precipitate forms.
g. Centrifuge at max speed for 10 minutes. A compact white pellet will form. Promptly proceed to the next step.
h. Insert a DNA Mini Column into a 2mL Collection tube.
i. Transfer the cleared supernatant from Step 7 by carefully aspirating it into the DNA Mini Column.
j. Centrifuge at max speed for 1 minute.
k. Discard the filtrate and reuse the collection tube.
l. Add 500μL HBC Buffer.
m. Centrifuge at max speed for 1 minute.
n. Discard the filtrate and reuse the collection tube.
o. Add 700μL DNA Wash Buffer.
p. Centrifuge at max speed for 1 minute.
q. Discard the filtrate and reuse the collection tube.
r. Repeat steps 15-17 for a second wash.
s. Centrifuge the empty DNA Mini Column matrix before elution. Residual ethanol may interfere with downstream applications.
t. Transfer the DNA Mini Column to a clean 1.5mL microcentrifuge tube.
u. Add 30-100μL sterile deionized water directly to the center of the column membrane.
v. Let sit at room temperature for 1 minute.
w. Centrifuge at max speed for 1 minute.
x. Store at -20℃
3. Yeast genome extraction:
a. Take yeast cell (no more than 5×107 cells), and centrifuge for 1 minute at 12000rpm.
b. Add 600μL sorbitol buffer to the bacteria,and then add 5μL lyticase. After blending, put it at 30℃ for 30 minutes, and centrifuge for 10 minutes at 4000rpm. After centrifugation, the supernatant was discarded and the precipitate was collected (To break down yeast cell walls).
c. Add 200μL GA buffer to the precipitate, and then resuspend it.
d. Add 20μL Proteinase K, and then blend the mixture.
e. Add 220μL GB buffer and blend it. Put it at 70 ℃ for 10minutes. The solution should become clear.
f. Add 220μL absolute ethyl alcohol. After blending it, floccose sediment will appear. Centrifuge if for short time.
g. Take the mixture to an adsorption column CB3, centrifuge for 30 seconds at 12000rpm. Discard the supernatant.
h. Add 500μL GD buffer, centrifuge for 30 seconds at 12000rpm. Discard the supernatant.
i. Add PW, centrifuge for 30 seconds at 12000rpm. Discard the supernatant.
j. Repeat the previous step.
k. Centrifuge for 2 minutes at 12000rpm. Discard the supernatant and dry CB3.
l. Transfer CB3 to a centrifuge tube. Add 50μL TE buffer. Centrifuge for 2 minutes at 12000rpm. Collect the solution.
b. Add 600μL sorbitol buffer to the bacteria,and then add 5μL lyticase. After blending, put it at 30℃ for 30 minutes, and centrifuge for 10 minutes at 4000rpm. After centrifugation, the supernatant was discarded and the precipitate was collected (To break down yeast cell walls).
c. Add 200μL GA buffer to the precipitate, and then resuspend it.
d. Add 20μL Proteinase K, and then blend the mixture.
e. Add 220μL GB buffer and blend it. Put it at 70 ℃ for 10minutes. The solution should become clear.
f. Add 220μL absolute ethyl alcohol. After blending it, floccose sediment will appear. Centrifuge if for short time.
g. Take the mixture to an adsorption column CB3, centrifuge for 30 seconds at 12000rpm. Discard the supernatant.
h. Add 500μL GD buffer, centrifuge for 30 seconds at 12000rpm. Discard the supernatant.
i. Add PW, centrifuge for 30 seconds at 12000rpm. Discard the supernatant.
j. Repeat the previous step.
k. Centrifuge for 2 minutes at 12000rpm. Discard the supernatant and dry CB3.
l. Transfer CB3 to a centrifuge tube. Add 50μL TE buffer. Centrifuge for 2 minutes at 12000rpm. Collect the solution.
5. Double enzyme digestion system

6. PCR reaction system


7. Reaction system of colony PCR

8. Overlap extension PCR:
a. Obtain the required enzymes, fragments and primers. The polymerase used in this experiment is PrimerSTAR HS (premix) produced by Takara Company.
b. Prepare PCR system. The system was composed of enzyme 50 μ L, front primer 2μL (0.2-0.3μM), back primer 2μL (0.2-0.3μM), two fragments per 1 L (concentration of 100ng/ μ L), sterile water 44μL, total system 100 L.

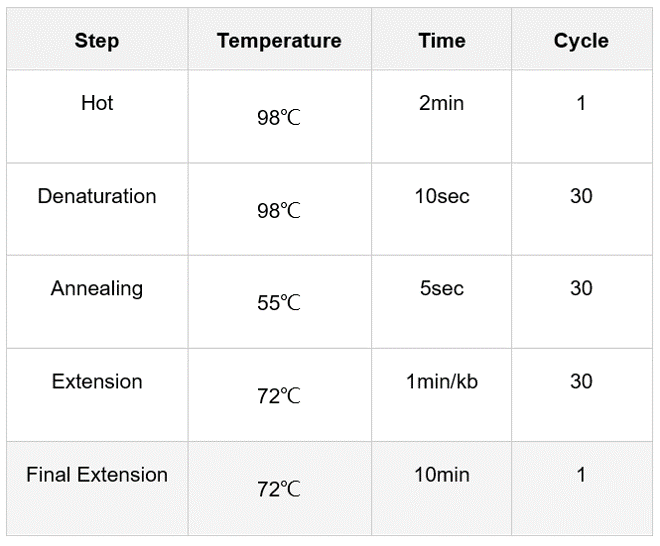
b. Prepare PCR system. The system was composed of enzyme 50 μ L, front primer 2μL (0.2-0.3μM), back primer 2μL (0.2-0.3μM), two fragments per 1 L (concentration of 100ng/ μ L), sterile water 44μL, total system 100 L.
9. Gel Extraction
a. Add 1 volume Binding buffer.
b. Incubate at 57℃ for 7 minutes until the gel has completely melted.
c. Insert a DNA Mini Column in a 2 mL Collection Tube.
d. Add all the DNA/agarose solution to the DNA Mini Column.
e. Add 300μL Binding buffer.
f. Centrifuge at max speed for 1 minute.
g. Discard the filtrate and reuse the collection tube.
h. Add 700μL SPW Wash Buffer.
i. Centrifuge at max speed for 1 minute.
j. Discard the filtrate and reuse the collection tube.
k. Repeat Steps 8-10.
l. Centrifuge the empty column for 2 minutes at the max speed to dry the column matrix.
m. Transfer the DNA Mini Column to a clean 1.5mL microcentrifuge tube.
n. Add 15-30μL deionized water directly to the center of the column membrane.
o. Let sit at room temperature for 2 minutes.
p. Centrifuge at max speed for 1 minute.
q. Store at -20℃.
b. Incubate at 57℃ for 7 minutes until the gel has completely melted.
c. Insert a DNA Mini Column in a 2 mL Collection Tube.
d. Add all the DNA/agarose solution to the DNA Mini Column.
e. Add 300μL Binding buffer.
f. Centrifuge at max speed for 1 minute.
g. Discard the filtrate and reuse the collection tube.
h. Add 700μL SPW Wash Buffer.
i. Centrifuge at max speed for 1 minute.
j. Discard the filtrate and reuse the collection tube.
k. Repeat Steps 8-10.
l. Centrifuge the empty column for 2 minutes at the max speed to dry the column matrix.
m. Transfer the DNA Mini Column to a clean 1.5mL microcentrifuge tube.
n. Add 15-30μL deionized water directly to the center of the column membrane.
o. Let sit at room temperature for 2 minutes.
p. Centrifuge at max speed for 1 minute.
q. Store at -20℃.
10. SDS-PAGE
a. Add 5x SDS-PAGE buffer.
b. Heat the sample under 95-100 ℃for 10 minutes.
c. Add 20μL sample to the DS-PAGE.
d. Add Coomassie Brilliant Blue and shake for several hours.
e. Discard Coomassie Brilliant Blue. Add destainer and shake it overnight.
b. Heat the sample under 95-100 ℃for 10 minutes.
c. Add 20μL sample to the DS-PAGE.
d. Add Coomassie Brilliant Blue and shake for several hours.
e. Discard Coomassie Brilliant Blue. Add destainer and shake it overnight.
12. Competent cell
a. The electric rotor should be washed 1 hour in advance and irradiated by ultraviolet lamp for 1 hour.
b. Precooling the electric rotor.
c. Select monoclonal yeast to 5mL YPD medium for overnight culture at 28℃. d. Inoculate 80-150μL solution into 100mL YPD.
e. Centrifuge at 5000rpm and 4℃ for 3 minutes. Discard the supernatant and collect the cell.
f. The cells were resuspended with 100mL precooled sterile deionized water. Centrifuge at 5000rpm and 4℃ for 3 minutes. Discard the supernatant and collect the cell into three tubes(30/30/30).
g. The cells were resuspended with 50mL precooled sterile deionized water. Centrifuge at 5000rpm and 4℃ for 3 minutes. Discard the supernatant and collect the cell into two tubes(25/25).
h. The cells were resuspended with 50mL precooled sterile potassium sorbate. Centrifuge at 5000rpm and 4℃ for 3 minutes. Discard the supernatant and collect the cell into one tube(25).
i. The cells were resuspended with 13mL precooled sterile potassium sorbate. Centrifuge at 5000rpm and 4℃ for 3 minutes. Discard the supernatant and collect the cell. j. Resuspend cells with 200-400μL precooled potassium sorbate (as little as possible).
b. Precooling the electric rotor.
c. Select monoclonal yeast to 5mL YPD medium for overnight culture at 28℃. d. Inoculate 80-150μL solution into 100mL YPD.
e. Centrifuge at 5000rpm and 4℃ for 3 minutes. Discard the supernatant and collect the cell.
f. The cells were resuspended with 100mL precooled sterile deionized water. Centrifuge at 5000rpm and 4℃ for 3 minutes. Discard the supernatant and collect the cell into three tubes(30/30/30).
g. The cells were resuspended with 50mL precooled sterile deionized water. Centrifuge at 5000rpm and 4℃ for 3 minutes. Discard the supernatant and collect the cell into two tubes(25/25).
h. The cells were resuspended with 50mL precooled sterile potassium sorbate. Centrifuge at 5000rpm and 4℃ for 3 minutes. Discard the supernatant and collect the cell into one tube(25).
i. The cells were resuspended with 13mL precooled sterile potassium sorbate. Centrifuge at 5000rpm and 4℃ for 3 minutes. Discard the supernatant and collect the cell. j. Resuspend cells with 200-400μL precooled potassium sorbate (as little as possible).
13. Transformation
a. Put competent cell TOP10 into the ice.
b. Add the DNA into TOP 10, gently resuspend.
c. Put the tube in the ice and incubate the cell for 30 minutes.
d. Take a heat shock (42°C) for 90 seconds. Promptly put the tube into ice.
e. Take an ice-bath for 3-5 minutes.
f. Add 200μL solution in a plate with antibiotic. Cultivate these bacteria overnight.
b. Add the DNA into TOP 10, gently resuspend.
c. Put the tube in the ice and incubate the cell for 30 minutes.
d. Take a heat shock (42°C) for 90 seconds. Promptly put the tube into ice.
e. Take an ice-bath for 3-5 minutes.
f. Add 200μL solution in a plate with antibiotic. Cultivate these bacteria overnight.
14. Electro transformation
a. Each tube is divided into 80-100μL competent cells, placed the tube into ice.
b. Add 7-15μL linearized plasmid and placed it on ice for 5 minutes.
c. Transfer to a precooled electric rotor and place it on ice for 5 minutes.
d. Shock it under the circumstance at 1500v, 5ms, 200Ω and 25μF.
e. Immediately add 1mL, 1 mol/L sorbitol. Mix it and transfer it into a sterile 1mL centrifuge tube.
f. 30℃ water-bath for 1-2 hours. Then adding 1mL YPD, culture for 1-2 hours.
g. Centrifuge at 12000rpm for 0.5 minutes. Remove the supernatant and add the rest of the solution in a MD plate with antibiotic. Cultivate the yeast overnight.
b. Add 7-15μL linearized plasmid and placed it on ice for 5 minutes.
c. Transfer to a precooled electric rotor and place it on ice for 5 minutes.
d. Shock it under the circumstance at 1500v, 5ms, 200Ω and 25μF.
e. Immediately add 1mL, 1 mol/L sorbitol. Mix it and transfer it into a sterile 1mL centrifuge tube.
f. 30℃ water-bath for 1-2 hours. Then adding 1mL YPD, culture for 1-2 hours.
g. Centrifuge at 12000rpm for 0.5 minutes. Remove the supernatant and add the rest of the solution in a MD plate with antibiotic. Cultivate the yeast overnight.
15. Assembly

a. Incubate at 50℃ for 15 minutes(1-2 fragments)、30 minutes(3 fragments)、60 minutes(4-5 fragments).
b. Store at -20℃.
16. Enzyme Activity
SLAC:
 VP:
VP:
 pelA:
pelA:
 Enzyme-labeled instrument
Enzyme-labeled instrument
a. Open the computer and double-click the SoftMax Pro v5 icon to enter the program.
b. Click the "Setting" button at the top of the plate panel.
 c. After entering the panel, select the leftmost square button "endpoint" and select "fluorescence" on the lower side of the panel, and click "OK".
c. After entering the panel, select the leftmost square button "endpoint" and select "fluorescence" on the lower side of the panel, and click "OK".
d. Fill the pop-up panel with excitation wavelength 460nm and emission wavelength 530nm.
e. After setting up, return to the "plate" page and click the "template" button.
f. Drag and drop the hole in the sample in the inner point of the box shown in the following illustration.
 g. Click on the "group" bar to select the “group”, and the blank control to select "blank"
g. Click on the "group" bar to select the “group”, and the blank control to select "blank"
h. To set the standard curve, select the "group" bar, click "New+", click "Standard" in "Column Format", and select the micro hole you want.
i. Click "series …" in the upper right corner of the dialog box. Enter the dialog box.
j. The unit, initial concentration and way to carry out the concentration gradient can be adjusted in turn. Select according to the requirements.
k. When measuring the sample to be tested, click on the "group" bar to select the grouping, click "Unknown" in "Column Format", and select the micro hole you want.
l. Wait for the data to come out, record the data.
VP:
pelA:
Enzyme-labeled instrumenta. Open the computer and double-click the SoftMax Pro v5 icon to enter the program.
b. Click the "Setting" button at the top of the plate panel.
c. After entering the panel, select the leftmost square button "endpoint" and select "fluorescence" on the lower side of the panel, and click "OK".d. Fill the pop-up panel with excitation wavelength 460nm and emission wavelength 530nm.
e. After setting up, return to the "plate" page and click the "template" button.
f. Drag and drop the hole in the sample in the inner point of the box shown in the following illustration.
g. Click on the "group" bar to select the “group”, and the blank control to select "blank"h. To set the standard curve, select the "group" bar, click "New+", click "Standard" in "Column Format", and select the micro hole you want.
i. Click "series …" in the upper right corner of the dialog box. Enter the dialog box.
j. The unit, initial concentration and way to carry out the concentration gradient can be adjusted in turn. Select according to the requirements.
k. When measuring the sample to be tested, click on the "group" bar to select the grouping, click "Unknown" in "Column Format", and select the micro hole you want.
l. Wait for the data to come out, record the data.
17. Component detection
Lignin:
a. Add 72% sulfuric acid 3mL to each tube containing lignocellulose, and stir 1minute with a glass rod until dissolve.
b. Water-bath the sample at 30℃ immediately for 1 hour. Stir once per 5minutes during the bath.
c. Wash the sample with 84mL sterile deionized water in a small triangular bottle. The sulfuric acid was diluted to 4%.
d. Seal the small triangular bottle with a sealing film. Heat preservation in 121℃ sterilization pot for 1 hour. Then cool to room temperature.
e. Take out the solution, filter it in a crucible, collect the filtrate, and repeatedly extract the filtrate with hot water until the filtrate is neutral.
f. Divide the filtrate into two parts.
g. The first part was used to determine acid-soluble lignin with approximately 3mL. The absorbance value of the filtrate at 320nm was recorded as A320. If the light absorption value is too large, it can be diluted properly. Double distilled water or 4% sulfuric acid was used as control.
h. Add calcium carbonate to the second part of the filtrate until neutral. Filter it with membrane, and do the high-performance liquid chromatography analysis. Determine the concentration of glucose, xylose and arabinose to calculate the content of cellulose and hemicellulose.
i. Wash the filter residue to neutral, and dry it in an oven at 105℃ for at least 4 hours. Cool it to room temperature, weighed as M1.
j. Ash the crucible with the sample in the muffle furnace at 575℃ for at least 24hours. After taking out, cool the sample to room temperature in the dryer and weighed as M2.
k. Acid Soluble Lignin(ASL)%=(A320×V(filtrate) ×n(dilute times)) / (ε×sample weight×b) ×100%
l. Acid Insoluble Lignin(AIL)%=(M1-M2) / 300mg
a. Add 72% sulfuric acid 3mL to each tube containing lignocellulose, and stir 1minute with a glass rod until dissolve.
b. Water-bath the sample at 30℃ immediately for 1 hour. Stir once per 5minutes during the bath.
c. Wash the sample with 84mL sterile deionized water in a small triangular bottle. The sulfuric acid was diluted to 4%.
d. Seal the small triangular bottle with a sealing film. Heat preservation in 121℃ sterilization pot for 1 hour. Then cool to room temperature.
e. Take out the solution, filter it in a crucible, collect the filtrate, and repeatedly extract the filtrate with hot water until the filtrate is neutral.
f. Divide the filtrate into two parts.
g. The first part was used to determine acid-soluble lignin with approximately 3mL. The absorbance value of the filtrate at 320nm was recorded as A320. If the light absorption value is too large, it can be diluted properly. Double distilled water or 4% sulfuric acid was used as control.
h. Add calcium carbonate to the second part of the filtrate until neutral. Filter it with membrane, and do the high-performance liquid chromatography analysis. Determine the concentration of glucose, xylose and arabinose to calculate the content of cellulose and hemicellulose.
i. Wash the filter residue to neutral, and dry it in an oven at 105℃ for at least 4 hours. Cool it to room temperature, weighed as M1.
j. Ash the crucible with the sample in the muffle furnace at 575℃ for at least 24hours. After taking out, cool the sample to room temperature in the dryer and weighed as M2.
k. Acid Soluble Lignin(ASL)%=(A320×V(filtrate) ×n(dilute times)) / (ε×sample weight×b) ×100%
l. Acid Insoluble Lignin(AIL)%=(M1-M2) / 300mg
Safety
Lab work:
The laboratory is a work environment where requires a lot of care, and safety is what we care most during our experiments. So we did a lot of preparation work before the experiment, the safety regulations are as below:
1.We received the strict safety training when joining the team, and we also have emphasized the personal hygiene before and during the experiments. Such as wearing lab coats and gloves no matter how simple the experiment is.【1】
2.In the course of our experiment, we have read the safety rules and policies carefully. We chose the safety reagent kit provided by the authorities.
3.We organized the study of Responsibilities page to ensure that everyone had a high level of responsibility for the whole project.
1.We received the strict safety training when joining the team, and we also have emphasized the personal hygiene before and during the experiments. Such as wearing lab coats and gloves no matter how simple the experiment is.【1】
2.In the course of our experiment, we have read the safety rules and policies carefully. We chose the safety reagent kit provided by the authorities.
3.We organized the study of Responsibilities page to ensure that everyone had a high level of responsibility for the whole project.
Project design:
1).In the selection of microbes we chose the pichia pastoris GS115 and E-coli Top 10, they're all risk group 1 microbes. And all of our gene fragments are also come from risk group 1 microbes and has no disease risk to humans and the environment. 【2】
2).Although we have only used E.coli and yeast cell, we did a critical safety assessment. Taking the possible impact on the public environment when disposing of waste cells into consideration, we sterilized the waste before we throw it away to guarantee the bacteria and fungus we used won’t be released out of the laboratory.
3).Our microbes all react in confined spaces which basically eliminating the risk of contamination in the process.
4).The product of each of our experiments offers no harm for any human, animal or plant, so it doesn’t matter even if we won’t have a suicide mechanism after the reaction.
5).HUST-China_Safety2019_Spreadsheet.xlsx[3]

2).Although we have only used E.coli and yeast cell, we did a critical safety assessment. Taking the possible impact on the public environment when disposing of waste cells into consideration, we sterilized the waste before we throw it away to guarantee the bacteria and fungus we used won’t be released out of the laboratory.
3).Our microbes all react in confined spaces which basically eliminating the risk of contamination in the process.
4).The product of each of our experiments offers no harm for any human, animal or plant, so it doesn’t matter even if we won’t have a suicide mechanism after the reaction.
5).HUST-China_Safety2019_Spreadsheet.xlsx[3]
Reference
[1] https://2019.igem.org/Safety/Risk_Groups
[2] Regulations on laboratory safety technology management of Huazhong university of science and technology.
[3] Canadian Pathogen Safety Data Sheets(PSDS)
[2] Regulations on laboratory safety technology management of Huazhong university of science and technology.
[3] Canadian Pathogen Safety Data Sheets(PSDS)
Notebook

