Team:Mines/Results


Binding:
We found that the sequence is too small to tell if it is present in the ligation with a pMAL vector. When trying to isolate the segment (which is about 75 bp long), the concentration of DNA was too small to be see in the gel. This can be seen in Figure 1.

Fig. 1- Digestion of the pUC19 vector with the binding insert with a 100 bp.
Based on the gels above, we saw that the segment was too small in the gel. We tried to use a different restriction endonuclease (BsaI) which would result in larger fragments. The expected gels from this digestion with and without the segment are shown in Figure 2.

Fig. 2 - Expected gels of size of fragments by cutting (a) the pMAL-p4x with the cadmium binding peptide (CBP) sequence and (b) the empty pMAL-p4x vector. The image was produced by the NEBCutter tool [1].
When running the gels, some of the ligations were negatives (wells a.4, b.5-b.7); however, the other wells were inconclusive as can be seen in Figure 3. We believe this was due to some interference in the digestion or that the insert was too short to be able to tell easily through restriction endonuclease digestion. The results could also be inconclusive due to bad samples as evident by the smearing.

Fig. 3 - Digestion of binding ligation to determine if the peptide insert is present
To find out the inconclusive results, we plan on replicating the ligation and digestion to see if there was interference in the procedure. We also plan to sequence the ligation to determine if the insert is in the vector without using restriction endonucleases.
Composite Binding Synthesis
We synthesized a new composite binding sequence that contains a tac promoter with the maltose binding protein and the binding sequence down stream. This sequence should translate the moltose binding protien with the binding sequence hanging off by a small linker. The purpose of the Maltose binding protein is to transport the binding protein to the periplasmic space in order to increase binding efficiency and lower the risk to the cell, and to stabilize the smaller protein. Primers for the sequence were made into to run PCR. Gel of PCR:
While the gel indicated some fragmentation, the concentration was found to be 325 ng/ml. This product and the pSB1C3 linearized plasmid were combined using Hifi Assembly. Subsequent transformation into chemical competent E. Coli resulted in no collony growth. This is was likely due to a storage error with the linearized plasmid.
Sensing:
The first test of the sensing plasmid from IDT did not show any difference in expression of the GFP between different concentrations of cadmium. The amount of fluorescence when adjusted to the amount of cells decreased with time. This could be explained due to the cells not being viable. The cells had a doubling time of about 9 hours which significantly longer than what is typical for E. coli. The slow growth may be due to the temperature of the well-plate reader being room temperature instead of 37°C. The cells were from an old plate possibly reducing the cell viability. Another possibility for the stunted growth is that the well on the plate did not provide optimal conditions for the cells. Some of the results from this test can be seen in Figure 4 and 5.

Fig. 4 - Fluorescence per cell for the first sensing test. The error bars represent 90% confidence.

Fig. 5 - Growth of cells for first sensing test. The error bars represent a 90% confidence interval
Some explanations of there being no difference in expression of the GFP between the different concentrations of cadmium are as followed:
- There is a nonspecific promoter ahead of the segment without a terminator.
- There is cadmium present in the media due to the media being in a glass container.
- There is promoter leakage from within the plasmid.
To eliminate these possible sources two routes were taken. The promoter, regulatory gene, an GFP sequence was attempted to be moved to the iGEM pSB1C3 backbone. The pSB1C3 backbone has 2 terminators before the section where the sequence would be added, However, the transformation was not successful.
The other route was using the IDT vector again, but using cells from a new plate, using media that was filter sterilized in plastic, and setting the plate reader to 37°C. These steps would make it so the media has minimal or no heavy metal contamination, that the new colonies were more likely to be viable, and use the optimal growth conditions. The results of the second test are shown in Figure 6. They show that all the colonies grew with about the same doubling time which was around 70 minutes. This is a higher value than the expected for E. coli, but does show viable growth.

Fig. 6 - doubling time for different cadmium concentrations for the second sensing test. The error bars represent a 90% confidence interval
This second test had an overflow of the fluorescence which may be because the emission and excitation wavelengths were too close to each other. Another explanation of the overflow reading that the GFP concentration was higher than expected due to more expression by the cells.
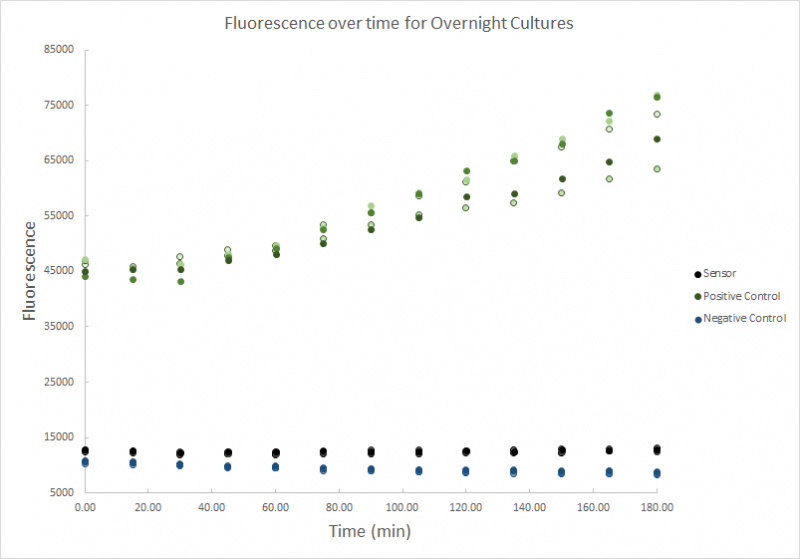
Fig. 7 - Fluorescence over time
The test was run again with the same concentrations and the results show the sensing was acting the same as the negative control. OD analysis of the cells indicates the doubling time of the cells was approximately 90 minutes. This can be seen in Fig. 8 The results indicate that the plasma may have been lost during storage.
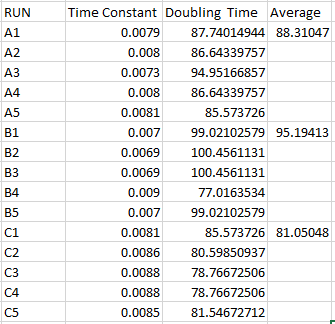
Fig. 8- Doubling Time regression
In the future we plan to:
- Move the insert into the iGEM plasmid
- Test sensing fluorescence and sensitivity
Further steps:
Beyond the short term goals, we are planning on longer term goals to improve and combine the different components. We plan to:
Use noncanonical amino acids to improve the affinity of the binding peptide with cadmium
Increase the cadmium resistance to the sensing plasmid.
Add the cadC promoter and regulatory gene in front of the binding protein fusion to selectively make the protein.
In the future the new component part can replace the tac promoter with a cadmium specific promoter to increase the specificity and reduce strain on the bacteria when no cadmium is present in the environment.
Reference
[1] Tamas Vincze, Janos Posfai, Richard J. Roberts; NEBcutter: a program to cleave DNA with restriction enzymes, Nucleic Acids Research , Volume 31, Issue 13, 1 July 2003, Pages 3688–3691, https://doi.org/10.1093/nar/gkg526