Team:Mines/Notebook

Mines Lab Notebook
During the spring semester troubleshooting the sensing model using a well plate reader was the primary objective. The first half of summer was spent on ligating, and transforming the original binding sequence into PUC19 and E. Coli. The second half of the summer was devoted to redesigning a functional part to avoid the ligation problems primarily due to the small length of sequence. Much of the school year was devoted to manipulating the new part, and characterizing a Cadmium promoter.
Wet Lab Documentation
Digested pMAL-insertion vectors and pMAL (negative control) with btsa1 RE:
| Component | Volume(uL) |
|---|---|
| pMAL (38 ng/uL) | 5 |
| 10X CutSmart Buffer | 5 |
| Btsa1 resctriction enzyme | 1 |
| Nuclease Free Water | 39 |
Reaction Conditions: 55°C incubation for 20 minutes. No heat inactivation used (might gel purify in the
future, this test is just to verify the presence of insert)
Used a 10% agarose gel at 120V for 50 minutes:
1 - GeneRuler 1 kb plus
2 - Empty
3 - pMAL no digestion, no insert
4 - pMAL w/ insert, no digestion (2.2)
5 - pMAL digested, no insert
6 - pMAL w/ insert (1.1), digested
7 - pMAL w/ insert (2.1), digested
Digested n-pMAL, 1.1-pMAL, 1.2-pMAL, and 2.1-pMAL with ApoI and BtaI
Used left over n-pMAL Btal digest
made concentrated digest (10ul insted of 50ul)
| Component | Volume (uL) |
|---|---|
| pMAL (38 ng/uL) OR pMAL -insert (unknown concentration) | 5 uL |
| 10X CutSmart Buffer | 1 uL |
| Btsa1, Apol, Restriction enzyme | 1 uL |
| Nuclease Free Water | 3 uL |
Ran Digest for 20 min, at 37°C for Apol and 55°C for Btal
inserted into gel, added 6 ul of loading dye to N-pMAL Btal digest, as there was around 40 ul of
solution to insert
Lane #
Ladder
Empty
n-pMAL #1 Btal digest
2.1-pMAL Btal digest
1.2-pMAL Btal digest
1.1-pMAL Btal digest
Empty
n-pMAL #1 Apo1 digest
2.1-pMAL ApoI digest
1.2-pMAL ApoI digest
1.1 -pMAL ApoI digest


Goals:
Finish Plates---done
Run PCR on binding protin sequence---done
run Topo protocol---done
transform Topo protocal---done
| Component | Volume (uL) |
|---|---|
| NEB 2x MM (M0270s) | 25 |
| M13 FP | 1 |
| M13 RP | 1 |
| IDT Oligo | 1 |
| Nuclease-Free Water | 22 |
PCR Conditions:
1. 94 °C 5 min
2. 94 °C 0:30
3. 45 °C 0:30
4. 72 °C 0:20
5. Repeated 2-4 30 cycles
5. 72 °C 7:00
6. 4 °C hold (was about 2 hours on hold before TOPO TA
Ran gel of PCR:
2% agarose gel, 110V, 40 minutes with 100 base Generule Ladder
Added 2 uL loading dye and 10 uL PCR product, loaded 10 uL of ladder and samples
Added 2 Lanes:
Generuler 100 bp ladder
---
PCR Replicate 1
---
PCR Replicate 2
PCR gel 3_31_2019.jpg
TOPO TA CLoning of bnd PCR:
Followed TOPO TA Cloning kit protocol by Thermo Fisher

| Volume (uL) | |
|---|---|
| PCR product | 4 |
| Salt solution | 1 |
| TOPO Vector | 1 |
Transformation
DH5-alpha
Followed TOPO TA Cloning protocol for One Shot DH5-alpha for transformation by Thermo Fisher
Did not use X-gaL solution on our plates
Tempature of water bath was not accurate, ranged from 40-45°C at 30 seconds
Added 900 uL of SOC to each solution instead of 250uL
added 100 uL of solution to plates with two controls for each sample
We have produced 6 plates and have 300 uL of solution left over for each sample
Plates are labeled 2.1 and 2.2, with the first number corrisponding to the sample number and the second
to plate number.. The controls are simply labled 2 for sample number
Made LB/ amp plates:
Added 20.0 grams agar to 1 L LB
Autoclaved on liquid setting (cover with foil and autoclave tape, loosen cap a bit so it doesn't explode
when applicable)
Let cool down to touchable temperature (when you can comfortably touch the flask for 10 seeconds)
Added 1 mL of 1000X ampicillin
Poured into plates and let dry. The lid was left slightly off to prevent condensation until it
cooled
Stored in fridge with lids on the bottom
Possible Errors in Cloning/Transformation
We never timed the mixture of the TOPO solution, we left it to run for more like ten minuets instead of
the perscribed 5. This could have lead to a degridation of the TOPO product
One shot cells were left on ice for a long time ~40 min, an not submerged, meaning they were posible too
warm to undergo heat shock
Heat shock was not an accurate tempature
we added 6 uL of TOPO product instead of 4, I cant think of a way that this would effect the
results.
Goals
Incubate lone colony from plate 1---done
restreak plates with left over solution---done
design plasmid for IDT to create
Icubate Colony
colony was removed from plate and placed in 5 mL of LB amp solution and left to incubate in shaker
overnight
Streak Plates
Added 300 ml of transformed bacteria to plates instead of 100, solution drained to cover of plates after
they were fliped- left overnight
Results from 5/31
The controls had growth but the LB amp plates exibited no growth exept for a lone colony. This colony is
most likely a mutation or forgen bacteria that managed to make it into the plate
Goals
make IDT vector
analyze lone bacteria colony that was growing
Analyze
Took the absorbance of the incubated ecoli solution- 1.287 at 600 nm, indicating growth
The plates showed no growth, indicating that the lone bacteria colony is most likely extranious.
To preserve the colony, the solution was replated and then diluted to two LB amp plates and two LB amp
solutions
The left over solution was then run through a miniprep to extract the plasmid from it and then run a
gell on the plasmid to see if it has the insert (ZymoPure Plasmid miniprep)
Plates produced a matt of colonies
Goals
Make IDT vector
Analize plates---done
rerun TOPO reaction on New PCR product----done
Analize
plates produced matts
PCR
We PCRed two products, the IDT Binding Vector OLIGO in tubes 1 and 2
| Volume (uL) | |
|---|---|
| Topo TA PCR product | 25 |
| Cut Smart | 5 |
| EcoRI | 1 |
| Denucleased water | 19 |
| Total | 50 |
| Component | Volume (uL) |
|---|---|
| NEB 2x MM (M0270s) | 25 |
| M13 FP | 1 |
| M13 RP | 1 |
| IDT Oligo | 1 |
| Nuclease-Free Water | 22 |
PCR steps were taken from MEB for the Taq 2 MM
95 at 1 min
30 cycles
95 at 30 sec
45 at 60 sec
68 at 20 sec
68 at 5 min
4 deg hold
All of 1-TOPO TA Mini 2115-a was used in the PCR the OLIGO and tube 2 were placed in box 2
| Component | Volume (uL) |
|---|---|
| NEB 2x MM (M0270s) | 25 |
| M13 FP | 1 |
| M13 RP | 1 |
| TOPO TA Mini | 22 |
| Nuclease-Free Water | 1 |
A 1.5 mL tube was prepared by the table above. Then placed in a 37 °C hot water bath for 30 minutes.
Blue White screening of the Lone Colony
Using 20 mg/ml of X-gal in DMSO, we plated one of the colonys on a LB/Amp plate
we heated the plate to 37°C for 30 min, we added 40 ul of x-gal soution to plate and incubated
......
We added 50 ul of Last Hope second gen to the plate in order to get more differentiation, and incubated
overnight at 37°C
| Volume (uL) | |
|---|---|
| PCR product | 4 |
| Salt solution | 1 |
| TOPO Vector | 1 |
Followed TOPO TA Cloning kit protocol by Thermo Fisher
left mixer on for 7 min
DH5-alpha
Followed TOPO TA Cloning protocol for One Shot DH5-alpha for transformation by Thermo Fisher
used X-gaL solution on 1 plate
3 plates were produced, 1 control, one with x gal, and one without
Gel Ran
lane 1 100 bp ladder
lane 3 new pcr product of TopoTA Cloning
lane 4 old pcr made
last hope minipreped and pcred.jpg
visibility was low, to bars of similar length were present, gel was stored in fridge for furthure
analysis.

Goals
Check results on new topo reaction---done
create freezer stocks---done
Created freezer stocks out of the Last Hope 2nd Gen
2ND TOPO Rxn
IMG_20190607_143407390.jpg
10 white colonys and 3 blue colonys were taken from this plate and inserted into 5 ml LB solutions to
incubate
these colonys are called A New Hope #1-10 and A New Hope Blue #1-3

Goals
Check A New Hope Colonys for insert
make more pMAL stock
Mini Prep of 4 white colonies and 2 blue (negative) colonies (New Hope batch)
1 ml of baterial culture was added to 1.5 ml tubes. Then the standard mini prep protocol was preformed.
Mini preps of White colonies 1-4, and Blue
colonies 1-2 were made.
pMAL Stock
pMAL plasmid was taken out and put into 5ml LB to grow
The 2nd TOPO Rxn B/W plate turned almost completely blue
Before:
2nd TOPO Rxn B_W plate.jpg
After
2nd Topo Rxn B_W plate next day.jpg
Mini Gel of Colonies Ran
Lanes
Gener Ruler 1 KB Ladder 10ul
---------
White 1 15 ul
White 2 15 ul
White 3 15 ul
White 4 15 ul
Blue 1 15 ul
Blue 2 15 ul
Conditions: 90 Volts 90 minutes


Goals
Insert Binding Portion into pMAL proten---in progress
test A New Hope minipreps for insert--- in progress
Topo To pMAL
Took pMAL precursor LB solution, put 100 uL on prewarmed LB/Amp plate, incubated at 37 for a day.
Minipreps
Made master mix
| Volume (uL) | |
|---|---|
| PCR product | 4 |
| Salt solution | 1 |
| TOPO Vector | 1 |
5 ul of master mix was pipeted into 6 centrifuge tubes, and 6 samples of minipreped DNA was inserted
into a seperate tube 5 ul each
samples were submerged in 37 degree water overnight
Gel Run of Digest
Lanes
100 Base Pair Ladder
2.
3. B1
4. B2 (Mostly Empty)
5. W1
6. W2
7. W3
8. W4
120 Volts for 45 minutes
Goals
Run gel of digested minipreps---done
Gel
standard load
very odd results
gel was soaked in ETBR soulution overnight


Goals
fugure out what the gel resutls mean
fix IDT Vector---done
Run Topo TA try 2 PCR products through gel---done
Gel
Possibly some combination of superfoiled, uncoiled, and cut
IDT vector
changes recorded in notes on vector
PCR
Standard loading conditions, no results
Goals
Run a gel of The Last Hope, The PCR that was inserted, and All the PCR products produced for TOPO TA try
2
Gel
Added 20 uL of solution to each lane, with an additional 5 ul of loading dye, added 25 ul of minipreped
and digested last hope-2118
Lane 1:Gene ruler 1kb
Lane 2: 100 bp ladder
Lane 3:
Lane 4: 2114-a
Lane 5: 2114-b
lane 6: 2118
Lane 7: Topo rxn 2, most likely used for TOPO insert, PCR
Lane 8: TOPO rxn PCR #2
All results are positive, even if they are faint. most concerning is the 2118 missing its larger back
bone, not even showing up in the gel. the lines also are closer to the 50 bp line than the 100 bp line,
when the insert should be about 120 bp
*****CORRECTION (6/7/19)*****
The ladder used in Lane 2 is the NEB Quickload 100 bp ladder (N0467S). The image of the ladder is below.
The bands are just above the 100 bp mark at the very bottom of the ladder, indicating that the DNA cut
from TOPO vector was successfully inserted. The error is likely due to referencing the Fisher 100 bp
ladder instead of the NEB quickload 100 bp ladder.



Goals
take swipe from pMAL to put in lb solution---no
pcr amplify sections of topo try 2 to check for insert---no
check gel for change---done
miniprep a last hope some of the pMAL precursor solution to prepare for insert---done
PCR amplify
m13 primers are missing: cannot continue
| Component | Volume (uL) |
|---|---|
| NEB 2x MM (M0270s) | 25 |
| M13 FP | 1 |
| M13 RP | 1 |
| TOPO TA Mini #2 | 22 |
| Nuclease-Free Water | 1 |
LB solution
not done
check gel
the stains have spread, lowering the resolution
miniprep
followed zymo protocal
produced two solutions: pmal and last
Individual colonies were selected from each plate and suspended in 5 ml of LB/Amp solution. Then
incubated at 37 C at 230 RPM. Two colonies from Pmal 1 were incubated.
6 freezer stocks were created using 600 ul of incubation solution, and 400 ul of 50% glycerol.
| Last Hope 1 | Growth on plate | Incubated over night | Successful incubation |
|---|---|---|---|
| Last Hope 2 | Growth on plate | Incubated over night | Successful incubation |
| Sensing 1 | Growth on plate | Incubated over night | Successful incubation |
| Sensing 2 | Growth on plate | Incubated over night | Successful incubation |
| Pmal 1 | Growth on plate | Incubated over night | Successful incubation |
| Pmal 2 | No growth | ||
| igem GFP | No growth | ||
| igem GFP | No growth |
The competent cells and ligation mixture were prepared, and a control of puc19 at 10 pg/ul was
also made with 5 ul of DNA added.
Competent cells were thawed on ice.
the cells were mixed with the plasmid as above.
The mixture was placed on ice for 10 minutes.
the cells were heat shocked for 30 seconds and placed back on ice.
950 ul of S.O.C. was added to the the solution
Solution was incubated at 37 C for 90 minutes shaking at 250 rpm.
Ampicillin selection plates were warmed
Plates of 10 and 100 ul of cells were spread.
Plates were incubated overnight at 37 C over night.
| Component | Volume |
|---|---|
| Chemically Competent cells | 50 ul |
| IDT Binding Plasmid (5ng/ul | 1 ul |
Goals:
PCR IDT Gene Block
Prmers with and without tag were synthesized from IDT
PCR with no tag primers preformed
PRC Settings:
Ran gell of pcr product of dna 2133
nanodrop concentration 325 ng/ul
Gel:
Lane 1: 100 bp Ladder (6 ul)
Lane 2: empty
Lane 3: 5 ul DNA 1 ml loading Dye
Results:
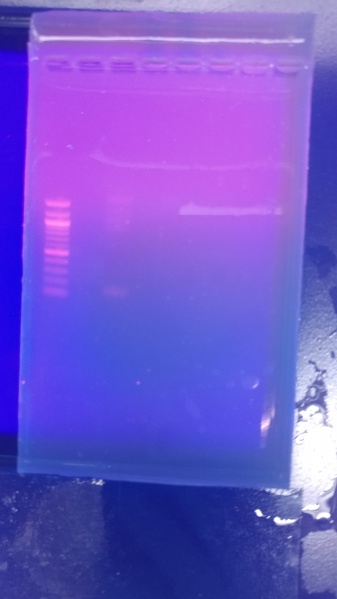
Hifi Assembly of IDT dna into Igem vector
Dna 2133 was diluted in a 1:1 ratio with dn water
4 ul of digested backbone was placed in pcr tube
added 1 ul of diluted dna 2133
5 ul of dn water added
10 ul of hifi master mix added
POSITIVE CONTROL
5 ul of puc 19 control added
5 ul of NEBuilder Positive control added
10 ul of hifi master mix
both samples put in pcr maching at 50 deg. C for 15 min
created chloramphencol plates
Protocol:
Mix:
10 g of Agar
12.5 g of Broth
500 mL of Deionized water
Autoclave
Add 12.5 mg of choramphencol after cooled to touch
Approximately 20 mL of solution was added to each plate. Plates were left for 30 minutes to solidify.
21 plates prepared 4 of normal lb for control purposes and the rest were chloramphenocol plates.
Goals
transfom and plate both positive control and DNA 2213 in ecoli
Rehydrolise and transform cad sensing protine for characterisation
Transform
nanodroped postive control and DNA 2213 (now DNA 2136)
Postive control- 1381 ng/ul
DNA 2136-1580 ng/ul
did math instead- got 18.1 ng/ul
need 1 pg -100 ng
5 ul of each sample required for transformation- assume postive control is similar
Rehydrolise
put 10 ul of nulcease free water to get 10 mg/ul
placed 1 ul of Cad protine for transformation
Transformation proctocol
unthawed three chemicaly compitent cells
c for positive control
b for DNA 2136
2h for cadA sensing
skiped dilution step, went straight to plating 10 ul and 100ul dillutions