
Scientific Background
Human caused climate change is the key issue in our era. Greenhouse gasses take a primary role in heating up the atmosphere. Animal agriculture is responsible for 18 % of anthropogenic greenhouse gas emissions, which is more than the combined exhaust of greenhouse gas of worldwide transport processes1.
Milk processing products account for 5-25 % of greenhouse gas emissions in Germany2, thus the problem is not only in animal farming, but also in the resulting products which also cause emissions and require resources.
The variety and benefits of cow's milk seem almost limitless and it is an essential element of nutrition and culture. So, the solution to reducing greenhouse gases can not be the total elimination of cow's milk.
The exact composition of milk has been known for a long time3. The ingredients that make the taste of milk unique are primarily proteins and lipids, as well as important enzymes. The remaining ingredients such as vitamins and minerals are already commonly synthesized without animals.
Approach / Aim
Our goal is the artificial production of milk proteins and fatty acids. The basis of our idea is to overproduce fatty acids and proteins using various synthetic biological, biotechnological and biochemical methods. Therefore, the gene sequences for the desired products were carefully selected and codon optimized.
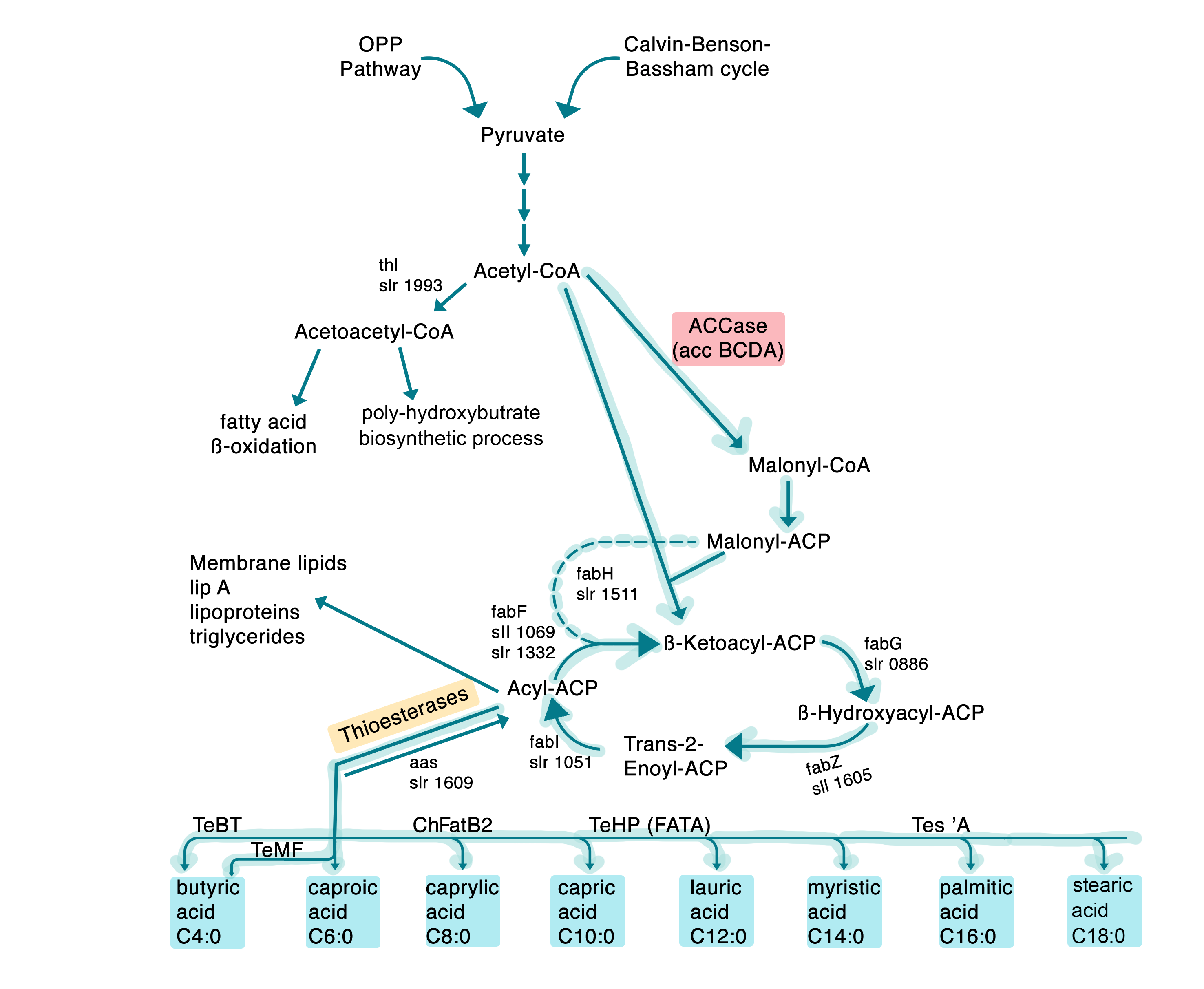
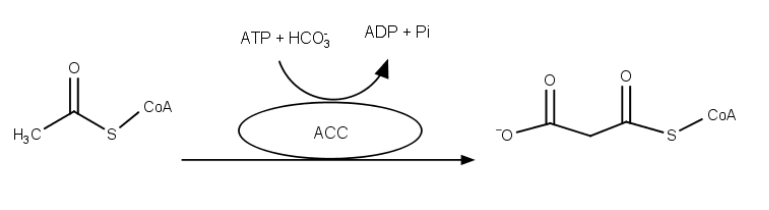
The starting point in fatty acid synthesis is acetyl-CoA, which is converted to malonyl-ACP by the Acetyl-CoA Carboxylase (ACC) complex . The conversion of acetyl-CoA to malonyl-ACP is the rate limiting factor in the production of fatty acids and is needed as a resource in the extension of these4.
We engineered the fatty acid synthesis pathway in several ways, starting with the overexpression of the ACC complex, decreasing the expression levels of unwanted enzymes and introducing heterologous thioesterases to terminate the fatty acid elongation at the desired length. The overexpression of the ACC complex was carried out in Escherichia coli to test the viability of this modification for fatty acid production, before expressing the complex in Synechocystis. The cyanobacterium Synechocystis sp. PCC 6803 is our host of choice for modular fatty acid production, because it does not release greenhouse gases, but performs oxygenic photosynthesis instead. In our project, it was used for experiments with CRISPRi-dCas9 and heterologous thioesterases.
For proteins, the main focus is on the caseins and the whey proteins. Those proteins represent the biggest part of protein components that can be found in the milk.
Similar to the fatty acid subproject, the protein sequences were optimized for overexpression in bacteria and subsequently expressed in various organisms. The overexpression and overproduction of both the fatty acids and proteins are the basis of our approach in the production of synthetic milk, so as a result of our work our aim is not to immediately produce a finished, consumable product, but the ability to continue and to build upon these results as basic steps for producing synthetic milk.
Cloning Strategies
| Name | used for | ORI | Copy number | Restistance | Size [bp] | Reference | Addgene |
|---|---|---|---|---|---|---|---|
| pET21a | ACC overexpression | pBR322 | around 15-20 | AmpR | 5443 | - | 69740-3 |
| pET22b | Casein production | ColE1 | 300-500 | AmpR | 5493 | - | 69744-3 |
| pBSMul1 (Jülich) | Casein production | f1 | 300-500 | AmpR, KanR | 7494 | 5 | - |
| NS4 | Fatty acid production | ColE1 | 300-500 | AmpR, KanR | 4958 | 6 | - |
| pSHDY | Fatty acid production | RSF1010 | 10-12 | CamR, SpecR, KanR | 9962 | 7 | - |
| pSNDY | Fatty acid production | RSF1010 | 10-12 | NatR | 10246 | 7 | - |
| pBbA6k | Biosensor, sensing free fatty acids | p15A | 20-30 | KanR | 3631 | - | 35291 |
| pBbB6c | Biosensor, sensing free fatty acids | pBBR1 | 5-10 | CamR | 3971 | - | 35356 |
The production of the main components of cow’s milk requires the use of synthetic biological approaches and modified microorganisms. The synthetically produced milk aims to achieve the same taste and nutritional value as cow’s milk, without using any animal products. Here, we mainly focused our project on the milk proteins, as well as long- and short-chain fatty acids.
In order to produce the milk proteins, different organisms were chosen, i.e. Escherichia coli, Bacillus subtilis, Pichia pastoris and Synechocystis sp. PCC 6803. The vectors pET22b, pBSMul1 and pSHDY/SNDY were used to create the intended molecular system. All used plasmids are high copy plasmids, as a maximum yield is wanted, except pSHDY/SNDY, which is a low copy plasmid.
For E. coli the strain BL21 (DE3) was chosen as an expression host. This strain represents a very convenient model system because of its beneficial biological traits. The BL21 (DE3) do not express a typical set up of endogenous proteases and encode the T7-Polymerase.
The applied vectors (pET22b, pBSMul1 and pSHDY) were amplified in transformed E. coli DH5ɑ using overnight cultures in LB-Amp (100 µg/ml) culture media, intending to isolate them in a high amount. The isolation was performed using commercial DNA isolation kits (Promega, Zymo Research). Afterwards, the vectors containing the genes ( ɑ-s1-, ɑ-s2-, β- and κ-casein, ɑ-lactalbumin and β-lactoglobulin ) were digested with the endonucleases BamHI and HindIII.
To purify the DNA, gel electrophoresis was performed using 1 % agarose in TAE buffer. The bands of correct DNA length were excised from the agarose gel and isolated by commercial DNA gel isolation kits (Promega, Zymo Research). The digested vectors were ligated with the inserts using sticky-end ligation with the T4 DNA ligase (NEB). After the ligation reaction, the solution was used to transform E. coli DH5ɑ, which was plated on LB-Agar with ampicillin (100 µg/ml). Colonies on the plate were tested with colony-PCR using primers that bind just outside the MCS.
Confirmed positive transformants were used to inoculate overnight cultures in LB medium with the appropriate antibiotics. The plasmid was prepped using a mini-prep kit (Zymo Research) and sent for sequencing (Sanger Sequencing by Eurofins Genomics). Samples showing successful sequencing results were used for transformation of E. coli BL21 (DE3) and subsequent overexpression experiments.
Bacillus subtilis was chosen as the primary production organism after the feedback from experts, as it can secrete proteins directly into the medium. This is advantageous for a food product, as fewer bacterial residues have to be removed during the purification. B. subtilis is better suited for this purpose as a gram positive bacterium, because it only has one cell membrane across which the protein has to be transported.
The research group of Prof. Dr. Karl-Erich Jaeger from the Institute of Molecular Enzyme Technology at Forschungszentrum Jülich provided plasmid variants with several different peptide signals, which facilitate secretion. The variants were all used, as the optimal peptide signal is different for each protein. Each milk protein was cloned into each plasmid variant, so the optimal peptide signal could be identified for each milk protein. They also provided the B. subtilis DB430 strain, which expresses fewer secreted proteases, increasing the stability of all secreted proteins8.
The pBSMul1 Plasmid was used with the following signal peptides: sslipA, SPYurl, SPNprE, SPPel and SPEpr. In this plasmid, protein expression is controlled by the constitutive HpaII promoter. pBSMul1 plasmids carry resistance cassettes against ampicillin and kanamycin. LB medium [20 g/L] with ampicillin [100 µg/ml] was used to grow overnight cultures of E. coli DH5ɑ to isolate the plasmids the following day. To purify the DNA, gel electrophoresis was performed using gels with 1 % agarose in TAE buffer. The bands of correct DNA length were cut out from the agarose gel and isolated by commercial DNA gel isolation kits (Zymo Research). Afterwards, plasmids and genes ( ɑ-s1-, ɑ-s2-, β- and κ-casein, ɑ-lactalbumin and β-lactoglobulin ) were digested with restriction enzymes NdeI and EcoRI. Each plasmid was ligated with each of the six inserts using the T4-Ligase (NEB). The plasmid product was then used for transformation of E. coli DH5ɑ and plated on LB-Agar with ampicillin [100 µg/ml]. Colonies on the plate were tested with colony PCR, positive transformant colonies were used to inoculate over-night cultures, which were used to isolate the plasmid. These plasmids were sent for Sanger Sequencing.
After positive sequencing results, naturally competent B. subtilis DB430 cells were transformed with the plasmid and plated on LB-agar containing kanamycin [25 µg/ml]. These colonies were confirmed to be positive transformants by colony-PCR before expression experiments were started. The genes encoding the milk proteins additionally contained a C-terminal 6x-His-tag to enable purification by affinity chromatography and immunodetection in Western blot.
Synechocystis sp. PCC 6803
For protein production in Synechocystis sp. PCC 6803, the conjugative broad-host range vector pSHDY7 was used. This vector is very convenient because it can be used in all bacteria capable of conjugation. It was provided by a supervisor with the promoter Pcpc560 expressing mVenus, which was codon-optimized for Synechocystis.
This vector was isolated from a E. coli DH5ɑ culture with a commercial miniprep kit by promega. pSHDY was digested with NheI and PstI and to purify the DNA, gel electrophoresis was performed using 1 % agarose in TAE buffer. The bands of correct DNA length were excised from the agarose gel and isolated by commercial DNA gel isolation kits (Machery-Nagel, Zymo Research).
The α-s1-casein and α-lactalbumin genes were ordered as complete gene fragments. Overlap extension PCRs were carried out to fuse the genes to the T1/T7 double terminator (BBa_0015). The resulting, elongated DNA fragment was digested with NheI and PstI as well and ligated into the pSDHY Pcpc560 vector using sticky-end ligation with T4-Ligase (NEB). This ligation mixture was used to transform E. coli DH5ɑ, yielding colonies on an LB-agar plate with spectinomycin. The colonies were checked for the insert by colony-PCR with gene-specific as well as non-specific primers, which bind outside the genes. Once they were confirmed as positive via sequencing of the isolated plasmid, they were used to inoculate an over-night culture. E. coli J539 with the RP4 plasmid was also inoculated. Synechocystis sp. PCC 6803 wild type liquid culture was grown to an OD750 of 0.6. The strains were used for a triparental mating process, which consisted first of the conjugation of the plasmids between the two E. coli strains and then the conjugation into Synechocystis on an BG11-agar plate containing 5 % LB-medium. The cells were mixed and plated out on a millipore HATF filter, allowing them to access the nutrients on the agar plate, but preventing cell movement. This keeps them in proximity to each other, increasing conjugation efficiency.
The Synechocystis cells were then carefully washed from the filter with liquid BG11 and transferred to a BG11-agar plate with spectinomycin to select for transconjugants. After 10-14 days, small colonies became visible, which were checked for the plasmid with the same colony-PCR as for the E. coli strains.
Beside the proteins in native cow’s milk, fatty acids are of major importance for the taste and texture. Thus, the industrial production of free fatty acids is a major goal to reach within “SynMylk”. The fatty acids are divided into short- (SCFA) and long-chain fatty acids (LCFA). The rate limiting step of the fatty acid biosynthesis is the conversion of Acetyl-CoA to Malonyl-CoA catalyzed by the Acetyl-CoA Carboxylase complex (ACC). Thus, overexpressing ACC will increase the level of free fatty acids in the organism.
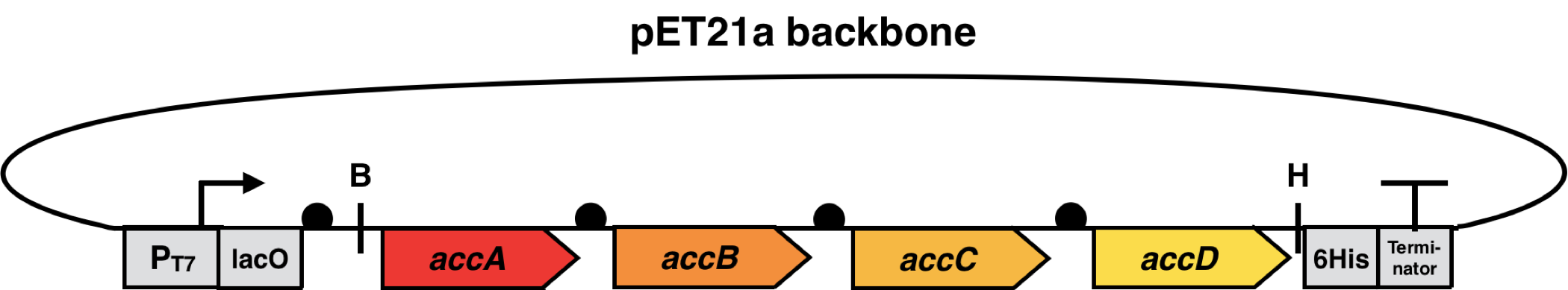
The genes encoding the ACC subunits of E. coli (accA, accB, accC and accD) were isolated by PCR from gDNA and cloned into a T7-promoter based vector containing AmpR (pET21a, Novagen) and the final plasmid was introduced into Escherichia coli BL21 (DE3). This E. coli strain is well known as an expression host with a deficiency in endogenous proteases. This is beneficial, because the overexpressed ACC protein complex needs to be protected from proteolytic digest to fulfill the conversion of Acetyl-CoA to Malonyl-CoA.
It is also important to mention that the antibiotic resistance (AmpR) and the origin of replication (pBR322) differs in comparison to the vectors used for the thioesterases and the biosensor. This enables a combination of ACC complex and thioesterase overexpression with a biosensor in one strain.
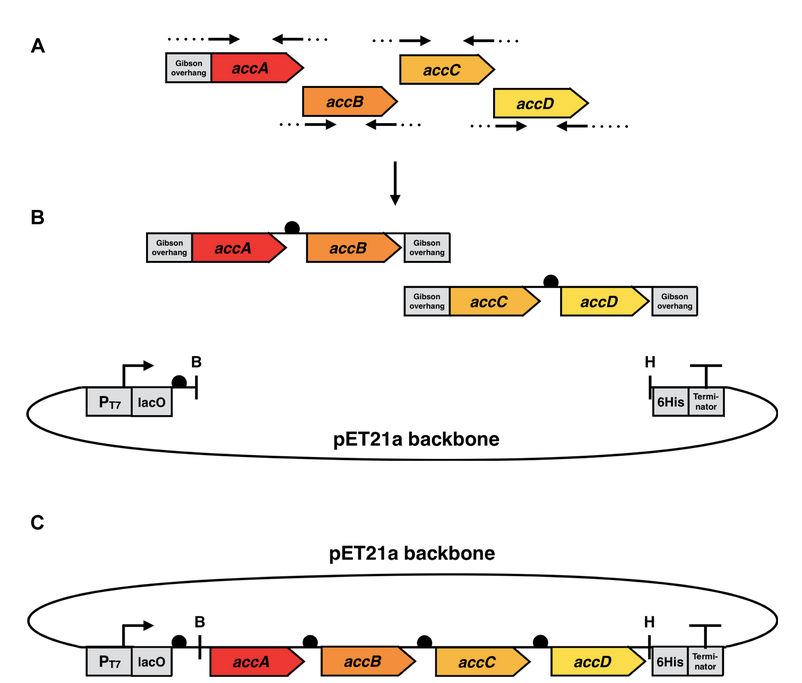
The cloning phase started with the amplification of the four acc-genes by polymerase chain reaction (PCR) using genomic DNA from E. coli K12 strain. After this, the single genes were fused to each other via overlap extension PCR using primers containing the Elowitz RBS (BBa_B0034). These PCR products were cloned into the pET21a vector by Gibson assembly and used for transformation of E. coli DH5ɑ and plated on LB-agar containing ampicillin (100 µg/ml). To control successful cloning, colony PCR and restriction digest of the constructs were performed. These internal quality controls indicated the successful insertion of accA-D genes into pET21a, prior to commercial Sanger sequencing of the construct.
The sequenced plasmid was used for transformation of E. coli BL21 (DE3) as the final production organism.
The expression is controlled by the T7 promoter, which enables transcription by T7 polymerase. The expression of this polymerase is inhibited by lacI and can be induced either by lactose or IPTG.
For the heterologous expression of thioesterases, Synechocystis sp. PCC 6803 wild type was used as main expression system and pSHDY7 as the vector of choice. This vector is very convenient because it is a broad range shuttle vector, which can be used in several organisms and therefore it is commonly used for conjugations, which is a method for transferring a plasmid from one organism into a different one.
This vector was isolated from a E. coli DH5ɑ culture with a commercial miniprep kit (promega). pSHDY was digested with EcoRI and PstI and to purify the DNA, gel electrophoresis was performed using 1 % agarose in TAE buffer. The bands of correct DNA length were excised from the agarose gel and isolated by commercial DNA gel isolation kits (Macherey-Nagel, Zymo Research).
The inserts were commercially synthesized as parts including a constitutive promoter BBa_J23119, a strong ribosome-binding-site (RBS*,BBa_K2924009) optimized for Synechocystis sp. PCC 680310, the CDS of the gene of interest and a double terminator BBa_B0015. With the method of Gibson assembly, the inserts were cloned into the digested vector as seen in Fig. 2.

E. coli DH5ɑ cells were transformed with the constructs, each containing a specific thioesterase, and plated on a plate with LB-agar and 25 µg/ml kanamycin. To ensure successful cloning, colony PCR was performed on the colonies. E. coli DH5ɑ strains containing the different thioesterase-encoding constructs were used for subsequent conjugation using the triparental mating method, where these plasmids were transferred from E. coli DH5ɑ to Synechocystis sp, as shown in Fig. 3.

Next to the pSHDY vector, the pSNDY vector was additionally used, which has the antibiotic resistance for nourseothricin, commercially available as clonNAT (Jena BioScience). It was isolated and digested with EcoRI and PstI, the same way as pSHDY. And all of the inserts containing the different thioesterases were cloned to the new vector by Gibson assembly. E. coli DH5ɑ cells were transformed with the constructs, each containing a specific thioesterase and plated on a plate with LB-Agar and 50 µg/ml clonNAT. To ensure successful cloning, colony PCR was performed on the colonies. Besides the constitutive promoter, a rhamnose inducible promoter was fused upstream of two of our thioesterases of interest, TeMF and ‘tesA, by overlap extension PCR, prior to cloning into a digested pSNDY vector variant containing the rhaS activator gene by Gibson assembly.

For an optimized fatty acid yield, a CRISPRi/dCas9-based knock-down-system was used. Four different off-target enzymes seem to be promising candidates and were chosen for downregulation to enable an increase in fatty acid yield. Additionally the mVenus was chosen for the proof-of-concept and also for this fluorescent protein a sgRNA were created. The gene-specific sgRNA variants were created with the tool “CRISPR Guides” by benchling and amplified by overlap extension PCR including the antibiotic resistance for kanamycin. The final overlap product was cloned into the EcoRI / PstI-digested NS4 backbone by Gibson assembly.
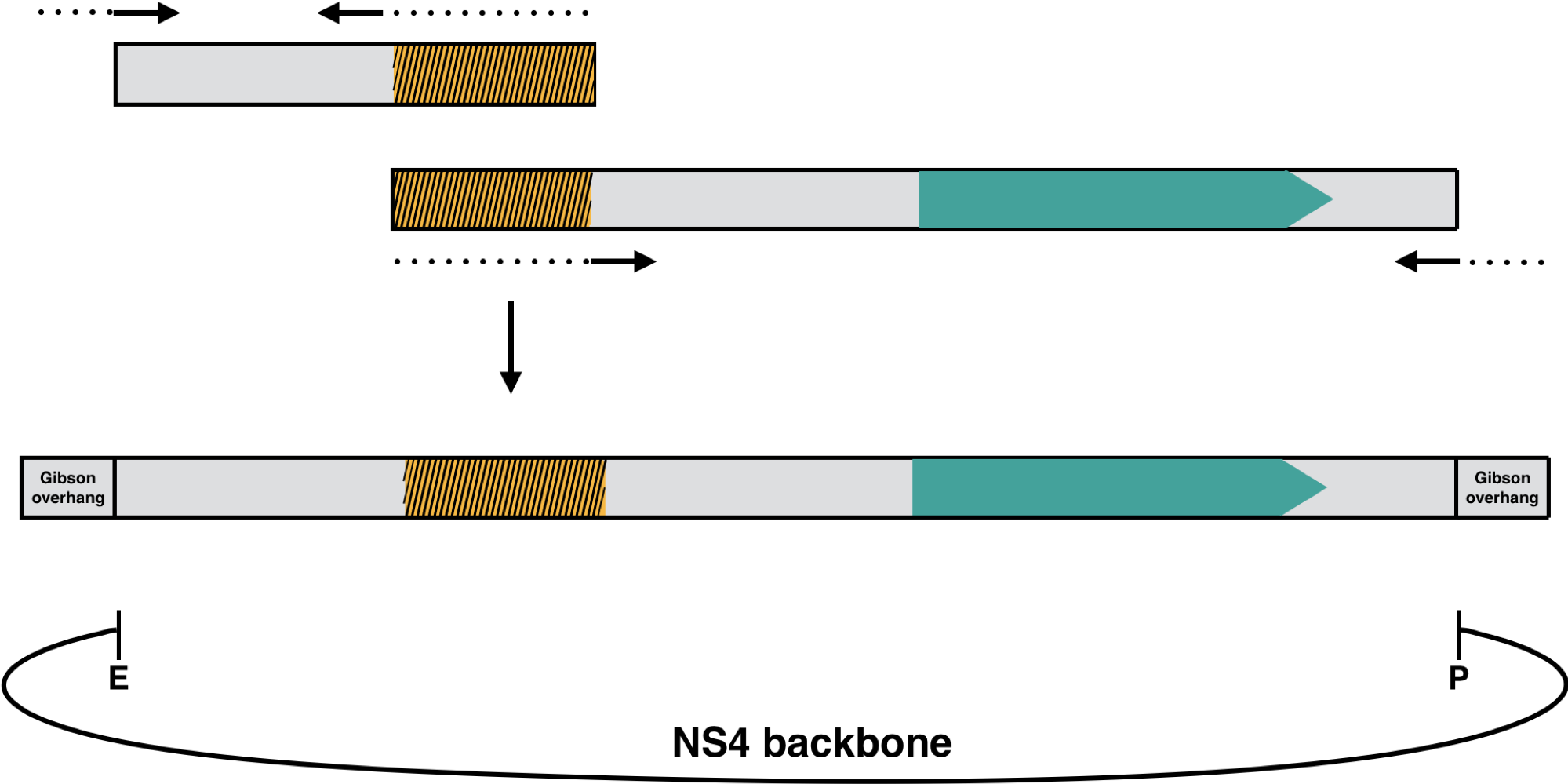
The NS4 plasmid, which contains the neutral site 4 (slr 2030 - 2031), and the Synechocystis sp. PCC 6803, which has integrated the dCas9 in its genome, were provided to us by Hudson et al. from Sweden and ensures the integration of the insert containing the antibiotic resistance and the sgRNA by homologous recombination into the homologous site NS4 of the genome of Synechocystis sp. PCC 6803. For this purpose, the ori ColE1 was picked for the plasmid, which allows a high copy number.
Synechocystis sp. PCC 6803 with the integrated dCas9 was transformed with 2 µg of the NS4 plasmid constructs and plated on a plate with BG11 and bacto-agar. A kanamycin gradient was obtained by carefully lifting the agar with a sterile spatula and adding 300 µL of kanamycin (1 mg/mL) underneath the agar. Plates were incubated until green colonies appeared at the edge where a lethal concentration of kanamycin could be observed. Green, isolated colonies were isolated from this plate and streaked out onto another plate with BG11 and bacto-agar containing gradually higher kanamycin. The concentration of kanamycin was increased with every next plate to ensure complete integration of the sgRNA-insert into the genome.
To ensure successful cloning and segregation, colony PCR was performed on the colonies.
With the pSNDY constructs containing a different antibiotic resistance than the NS4-dCas9 strains, it was possible to use both constructs in one organism. For this purpose, the Synechocystis knock-down strains received a pSNDY construct by transferring the plasmid by triparental mating.
For a fast detection of free fatty acids, a biosensor with different reporter genes was designed and ordered as gene fragments from IDT. Since the backbones pBbB6c11 and pBbA6k11 were used, the parts containing different promoters and reporter genes were designed using the BglBrick standard, a modified variant of the BioBrick standard. The restriction enzymes EcoRI and XhoI were used for digesting the backbone and inserts and the parts were ligated into pBbB6c and pBbA6k variants lacking their original inducible promoters.
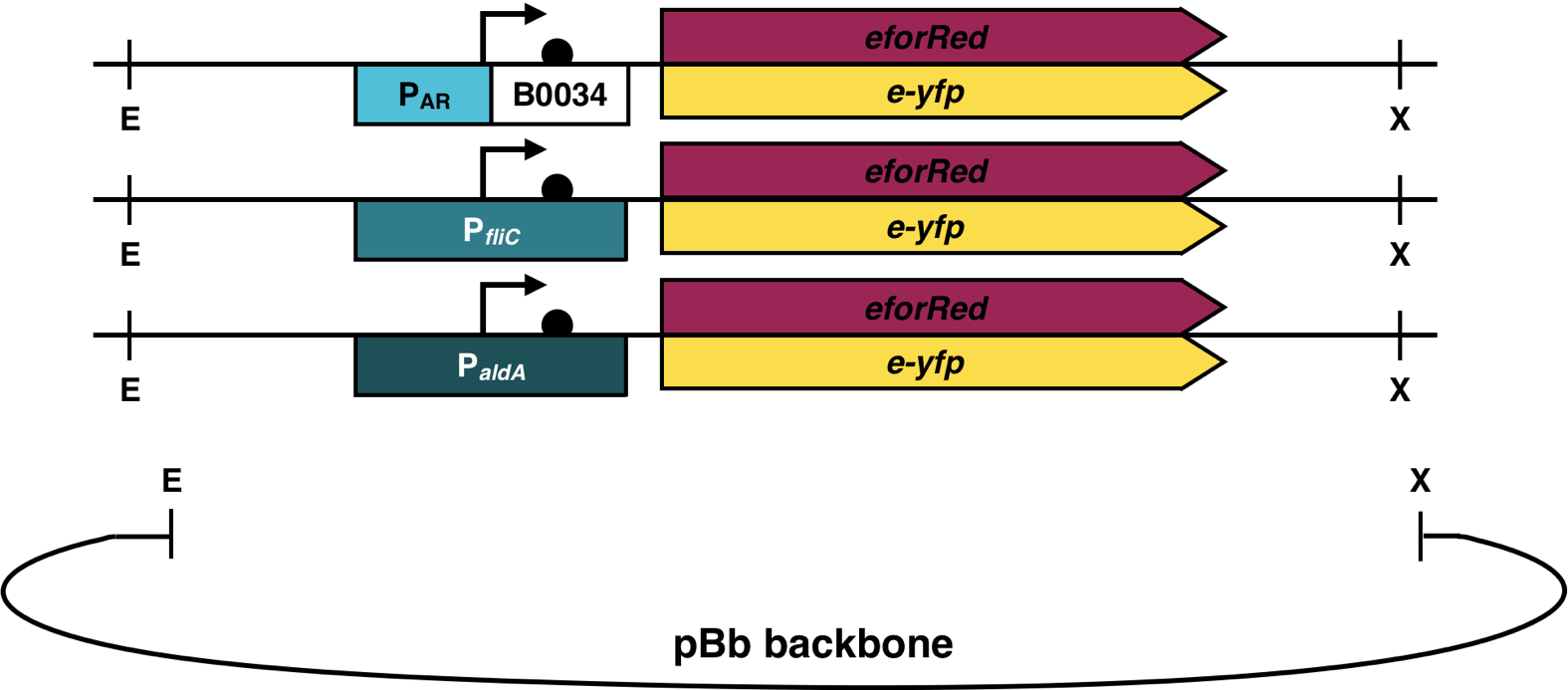
The backbone pBbB6c11 has the antibiotic resistance chloramphenicol with the ori pBBR1 and the backbone pBbA6k11 has the antibiotic resistance kanamycin with the ori p15A. The plasmids were used for transformation of E. coli DH5ɑ and plated on LB-agar containing appropriate antibiotics. The colonies were confirmed by colony PCR and positive colonies were inoculated and grown in LB medium with chloramphenicol or kanamycin and the plasmid was isolated for subsequent Sanger sequencing. The isolation of the plasmid was performed using a commercial plasmid preparation kit (Promega). The sequenced plasmids were used for transformation of E. coli Top10F and E. coli BL21 (DE3) and plated on LB-Agar with the appropriate antibiotic. The decision for these antibiotics and ori was made to combine the biosensor with the plasmids of the thioesterases and the Acetyl-CoA carboxylase complex. For this, the plasmids of the different parts were combined in many transformation combinations in E. coli BL21 (DE3).
| Addition | Promoter | Reporter Gene | Antibiotic restistance |
|---|---|---|---|
| - | aldA | eYFP | Kanamycin |
| - | filCA | eYFP | Kanamycin |
| - | AR | RFP | Chloramphenicol |
| - | AR | sfGFP | Chloramphenicol |
| - | AR | amilCP | Chlorampheniol |
| HP | aldA | eYFP | Kanamycin, CloNat |
| tesA | aldA | eYFP | Kanamycin, CloNat |
| MF | fliC | eYFP | Kanamycin, CloNat |
| BT | fliC | eYFP | Kanamcin, CloNat |
| accABCD | aldA | eYFP | Kanamycin, Ampicillin |
| accABCD | fliC | eYFP | Kanamycin, Ampicillin, CloNat |
| accABCD + HP | aldA | eYFP | Kanamycin, Ampicillin, CloNat |
| accABCD + MF | fliC | eYFP | Kanamycin, Ampicillin, CloNat |
| accABCD + BT | fliC | eYFP | Kanamycin, Ampicillin, CloNat |
| accABCD + tesA | aldA | eYFP | Kanamycin, Ampicillin, CloNat |
| accABCD + tesA | AR | sfGFP | Chloramphenicol, Nicol, Ampicillin, CloNat |
The transformations were done to test the activity of the biosensor in vivo to scan for increased production of free fatty acids by overexpression of the Acetyl-CoA carboxylase complex and the thioesterases.


The double and triple transformation were made to test the activity of the biosensor in vivo to scan if the production of free fatty acids by the Acetyl-CoA carboxylase complex and the thioesterases work. This setup of plasmids, oris and antibiotics made it able to have a high modularity for the possible combination of the biosensor.
Experimental Design
The original cow’s milk contains 4.2 % fat13 - among other main components. These fats mainly consist of fatty acids, which come in average chain lengths between four, sixteen or eighteen carbon-atoms. The fat is responsible for the taste, consistency and texture of the milk. To obtain the most native-like cow’s milk without actually using cow-derived ingredients, we focused on synthesizing the substantially needed fatty acids. These fatty acids are not commonly synthesized, because common methods of production is not convenient. But they are essential for the taste and appearance of the milk. For example, butyric acid (C4:0) contributes to a major part to the consistency of the milk.
For this purpose, we chose five different thioesterases, as indicated in table 1. These are known to produce the required short-chain fatty acids, which include C4:0, medium-chain fatty acids including C6:0 to C12:0 and long-chain fatty acids C16:0 and C18:0, as seen in the table. The thioesterases are derived from different organisms. Thioesterases terminate the elongation of fatty acids due to their hydrolytic activity, thereby catalyzing the synthesis of free fatty acids. Each of the five thioesterases chosen terminates the elongation at specific chain lengths.
| sample ID | organism | chain lenght | reference | Genkbank ID |
|---|---|---|---|---|
| ChFatB2 | Cuphea hookeriana | C8:0 and C10:0 | Dehesh, Katayoon, et al.14 | U39834 |
| TeMF | Marvinbryantia formatexigens | C4:0, C6:0 and C8:0 | Jawed, Kamran, et al.15 | EET61113.1 |
| TeBT | Bacteroides thetaiotaomicron | C4:0 | Liu, Xiping, et al.16 | AAO77182.1 |
| TeHP | Haematococcus pluvialis | C16:0 and C18:0 | Lei, Anping, et al.17 | HM560034 |
| ´TesA | Escherichia coli | C16:0 and C18:0 | Cho, Hyeseon, and John E. Cronan18 | 94512720 |
Experimental Design
The thioesterase genes were from ebi.ac.uk or ncbi.nlm.nih.gov and codon-optimized for Synechocystis sp. PCC 6803. The promoter BBa_J23119, the RBS* BBa_K2924009, a synthetic strong RBS designed for Synechocystis10, and the double terminator BBa_B0015 were added up- and downstream to each thioesterase CDS and synthesized by Doulix, as shown in Fig. 10. The promoter BBa_J23119 is the strongest constitutive member of the J23 promoter family. These parts were each cloned into the pSHDY plasmid, which was provided by Behle et al.7. The pSHDY plasmid is an RSF1010-based, low-copy self-replicating vector derived from pVZ32119 and has a broad host range, which enables the conjugative transfer from E. coli to cyanobacteria and other microorganisms. For the uptake of the plasmid, the triparental mating method was used. The thioesterases teMF and ‘tesA were cloned with Prha, a rhamnose inducible promoter, instead of the constitutive promoter BBa_J23119. This promoter is already present in the registry as BBa_K914003 and was submitted by the team of Paris Bettencourt 2012. However, the sequence used by us differs from this part by one bp, likely modified by Paris Bettencourt 2012 to exclude the illegal BioBrick restriction site EcoRI. Since this promoter was already used successfully by Yao et al.6 and Behle et al.7 in Synechocystis, and the base pair exchange is close to the region relevant for promoter functionality, we chose to use the original promoter containing the EcoRI site. The final organism targeted for overexpression of these constructs is Synechocystis sp. PCC 6803.

To improve the yield of free fatty acids, knock-downs of several genes involving in the metabolism of Synechocystis sp. PCC 6803 were planned and created. These knockdown constructs contain a kanamycin resistance and Synechocystis sp. PCC 6803 contains the dCas9 gene with a spectinomycin resistance. Therefore, the diverse thioesterase genes were additionally cloned via Gibson Cloning into the pSNDY backbone, which contains a nourseothricin (clonNAT) resistance, present in the iGEM registry as BBa_K2799001. By using the clonNAT resistance, it is possible to combine the heterologous overexpression of thioesterases and the knockdown of different genes, in one strain, potentially yielding optimized production strains. Besides just combining the knockdown constructs with the thioesterases, it is also conceivable to combine the accBCDA BBa_K2924021, which contains the antibiotic resistance for ampicillin. The accBCDA is, as shown in Fig. 9, also involved in the fatty acid metabolism as the rate-limiting step for fatty acid synthesis. Also possible is combining diverse biosensor with the thioesterases (for e.g. BBa_K2924019 and BBa_K2924020 ; they contain the antibiotic resistance for chloramphenicol). Biosensors are genetic switches which respond to small molecules, such as fatty acids, usually in the form of a simple output-signal as fluorescence. This enables the detection and quantification of the produced free fatty acids, without using a expensive method like GC-MS. All of these constructs can also be combined and expressed in one single strain due to the overall different antibiotic resistances and origins of replication for efficiently producing a high yield of fatty acids and detecting them simple and fast by visible and quantifiable response.
CRISPR Knockdown
The primary aim was to synthesize short-chain fatty acids through heterologous overexpression of various enzymes which are involved in fatty acid biosynthesis, using the unicellular cyanobacterium Synechocystis sp. PCC 6803 as the expression host. Since changing or extending the metabolic network of the cell can have drastic effects on cell viability, trade-offs between producing the desired products and the cell's own production of substances could occur. To reach higher yields, possible knock-outs of diverse enzymes involved in off-target reactions were shown to be applicable. Since some of those knock-outs can be lethal for the organism, which can be modeled by a flux balance analysis, the idea of down-regulating the expression of major enzymes in the related pathways occurred to us. A CRISPR/dCas9-System, published by Yao et al.6 and kindly provided by Dr. Elton P. Hudson (KTH, Sweden), is the method of choice6. A catalytically inactive Cas9 (dCas9) was used, which is able to bind to the target DNA, but does not introduce a double strand break, thereby blocking the RNA-Polymerase and subsequent transcription of the gene of interest (Fig. 11).
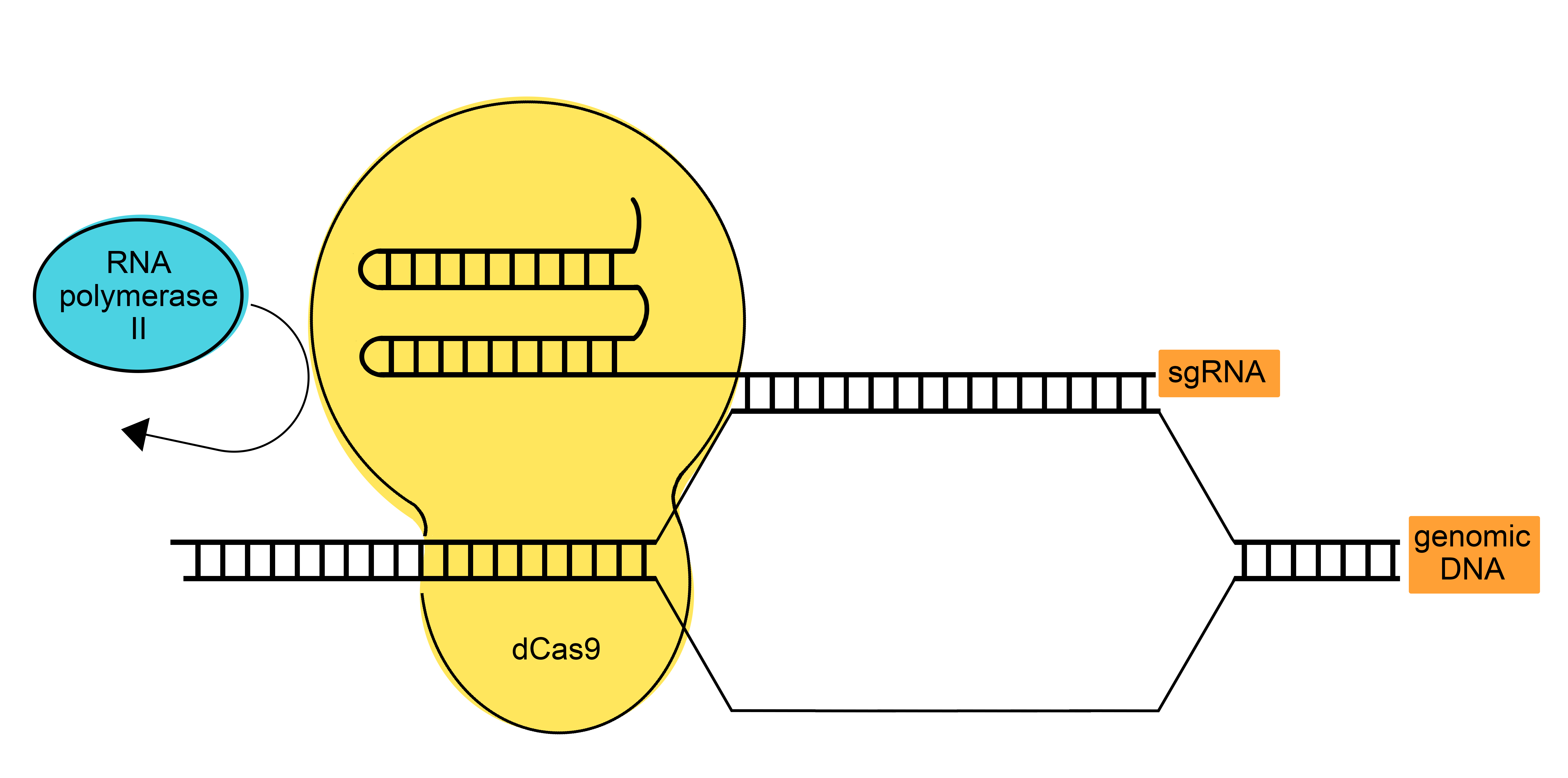
DCas9 finds its target due to a gene-specific small guide RNA (sgRNA), which forms a complex together with dCas9 that binds to the complementary DNA strand. Four target enzymes of the fatty acid metabolism, the long-chain-fatty-acid CoA ligase, enoyl-[acyl-carrier-protein] reductase, β ketoacyl-acyl carrier protein synthase and the acetyl coenzyme A acetyltransferase (thiolase), which are shown in red in Fig. 4, were chosen for down-regulation. From these targets, sgRNAs were designed using the the tool “CRISPR Guides” on benchling, choosing candidate sgRNAs which bind at the start of the desired genes, for the dCas9-System.
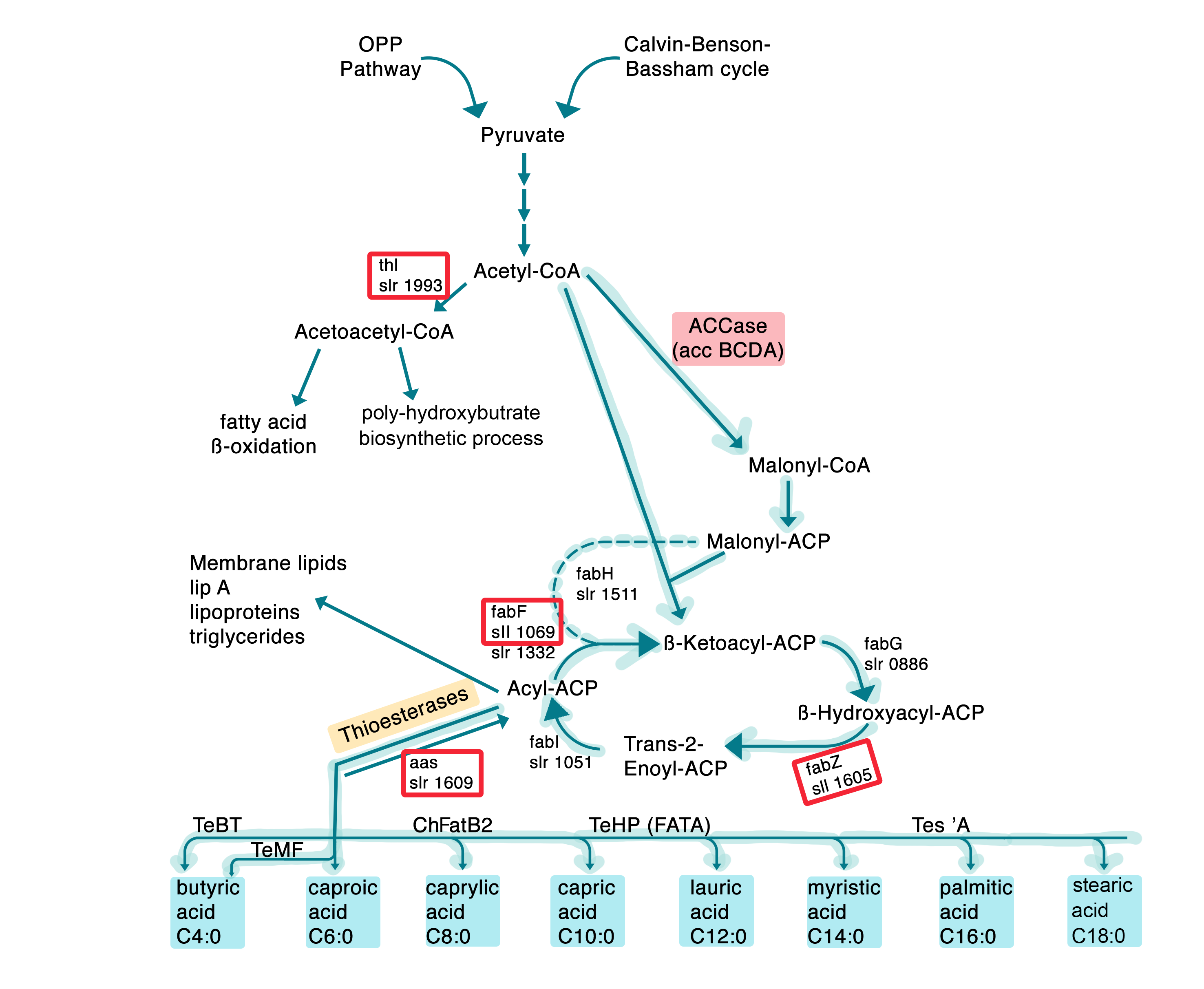
These guide RNAs were synthesized with specific primers by overlap extension PCR and further cloned via Gibson assembly into a high-copy plasmid for genomic integration into the slr2030-slr2031 neutral site 4 (NS4)6, which was termed NS4 plasmid (Fig. 13). NS4 stands for the neutral site 4, which is homologous to a certain region in the genome of Synechocystis sp. PCC 6803 and with this it can be inserted into the genome (Fig. 14).
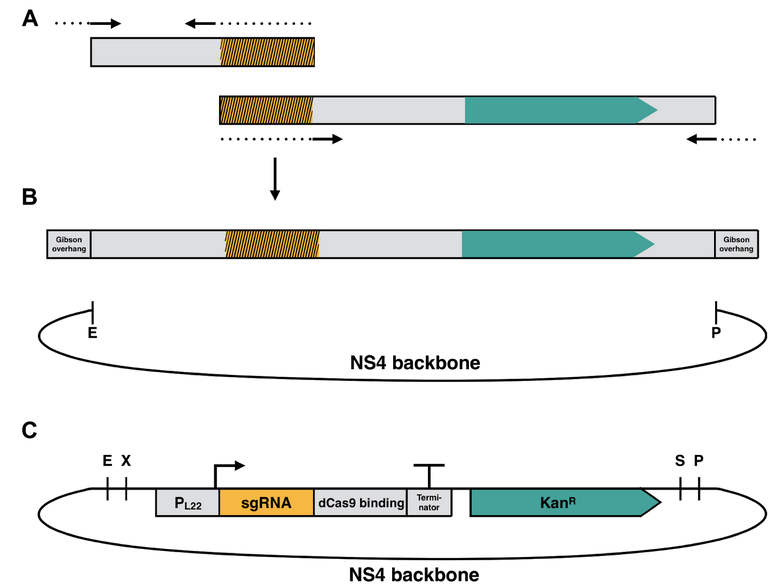

Besides the guide RNA, the insert contains resistance for kanamycin as a selection marker, which enables selection of positive clones after transformation. The cyanobacterium Synechocystis sp. PCC 6803 contains a with anhydroTetracycline (aTc) inducible dCas9 at the psbA neutral site. This includes a spectinomycin and a tetR cassette6. Furthermore this strain is capable of taking up foreign DNA and is able to integrate it into its genome, which makes them interesting targets for mutagenesis21. Also, the fully sequenced genome of Synechocystis sp. PCC 6803 is a major benefit of this strain, which makes it very convenient to work with it. Synechocystis sp. PCC 6803 contains three CRISPR/Cas - systems, one type 1 and two type 3. Furthermore, the proposed strain also contains the heterologous type 2 dCas9 - system6.
Fatty acid and lipid metabolism have been studied in detail throughout the past decades. The underlying mechanisms, genes and enzymes involved in fatty acid biosynthesis, degradation and regulation, as well as global regulation on the metabolic level have been studied extensively in microbial species, plants and animals. In order to effectively utilize this information, synthetic biology can be incredibly helpful to further elucidate and apply genetic tools.
One approach of synthetic biology entails engineering biosensors. Biosensors are genetic switches which respond to small molecules in a dose-dependent manner, usually in the form of a simple output signal, such as fluorescence or chromoproteins. This enables detection and quantification of these small molecules without costly extraction and quantification methods.
The quantification of fatty acids is currently done via GC-MS, which is both costly and often inaccessible to iGEM teams around the world. Therefore, the aim is to explore different genetic building blocks and determine their ability to sense the presence of different fatty acids in a dose-dependent manner. This would represent a cost-effective tool for quick proof-of-concept fatty acid production experiments prior to quantification with the GC-MS, with the potential for high-throughput screening of fatty acid production microbial strains.
Experimental Design
For the biosensor with the promoters PAR, PLR and PfadBA, a FadR-dependent reporter system was used11. The transcription is blocked by the regulator FadR when fatty acids are not available and the reporter gene can not be transcribed. After the production of fatty acids, the regulator FadR is released from the promoter region and bound to the fatty acids and the reporter gene can now be transcribed (Fig. 15).

Different promoters with different sensitivity for specific fatty acid chain lengths were used. The promoter PAR::CDS is sensitive for long chain fatty acids. This promoter was published for oleic acid (C18:1) and the iGEM team from Barcelona 2018 tested it with palmitic acid (C16:0). Therefore, we tested all relevant fatty acids with various concentrations.
For the myristic acid (C14:0) and palmitic acid (C16:0) we used the promoter PfadBA:CDS. With the promoter PfliC:CDS we are able to measure butyric acid (C4:0) and the promoter PaldA:CDS can also detect oleic acid (C18:1)22. These promoters were combined with eYFP as a fluorescent reporter gene and eforRed as a chromoprotein for absorption measurement, because not every iGEM team may be able to measure fluorescence.
The biosensors can be further combined with other parts of the present project, like the thioesterases for the production of SCFA and the overproduction of the ACC complex, the rate limiting enzyme complex of the fatty acid synthesis. Additional measurements between in vivo production and adding the fatty acids externally to the supernatant will further elucidate the level of detection of the proposed biosensor. All measurements were done in Escherichia coli bacteria, because of the fast growth rate and the simple laboratory conditions required by these particular class organisms.
The amount of possible carbon chain combinations leads to the existence of approx. 400 different fatty acid variants in the milk fat. In the project, we mainly focused on 4 LCFAs which are the most abundant: The 3 saturated fatty acids myristic acid (C14:0), palmitic acid (C16:0) and stearic acid (C18:0), and finally the monounsaturated omega-9 oleic fatty acid (C18:1)13, 23. To produce the aforementioned fatty acids, one of the host organisms we chose was Escherichia coli. This model organism already has a fatty acid biosynthesis pathway (Fig. 16), where the desired fatty acids chain lengths are produced. In order to obtain increased amounts of free fatty acids, the rate limiting steps of fatty acid biosynthesis were determined by modelling. Since one of the main bottlenecks for the biosynthesis of fatty acids is the conversion of Acetyl-CoA to Malonyl-CoA, the four genes of the Acetyl-CoA Carboxylase complex (ACC), which catalyzes this reaction, were cloned and overexpressed.
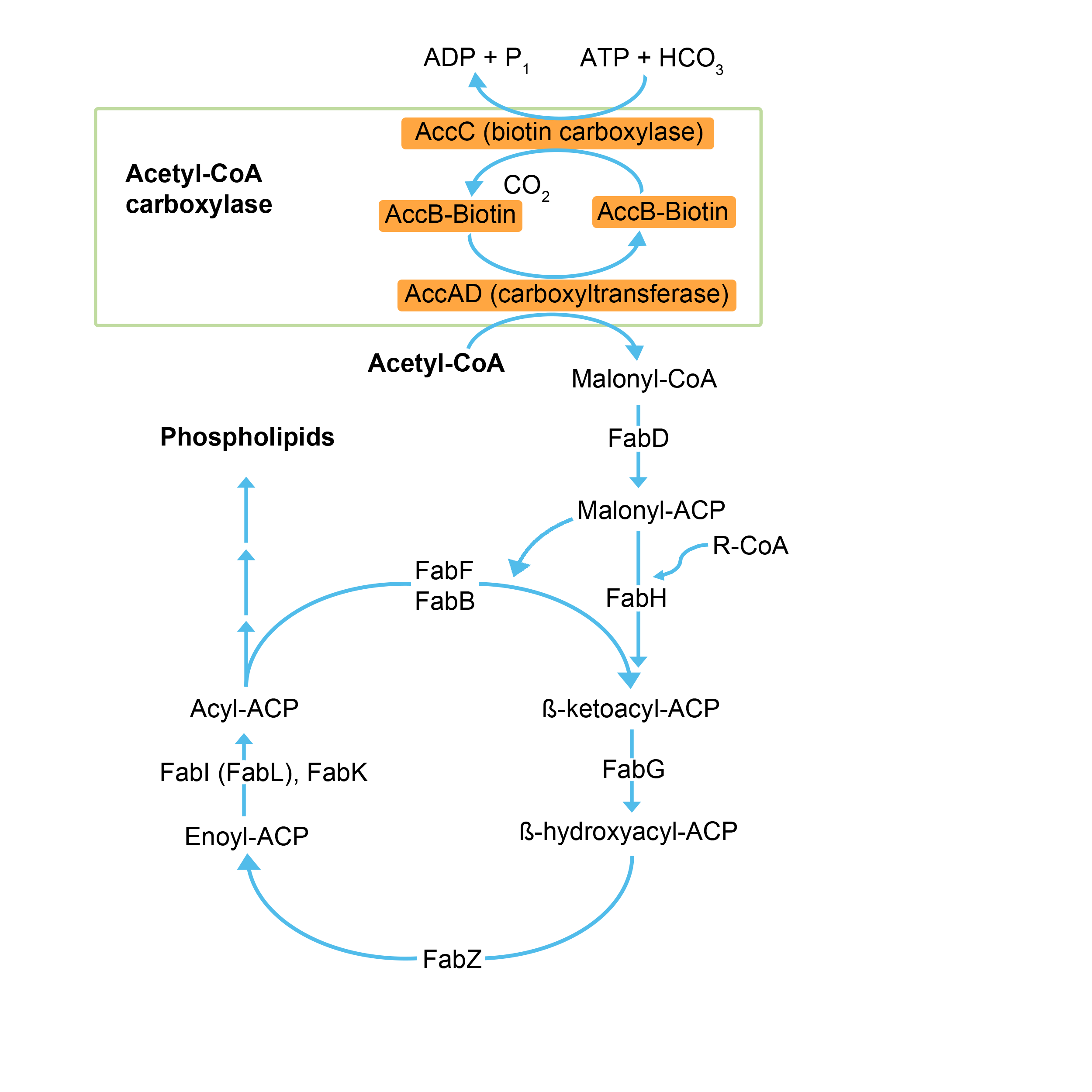
ACC in E. coli is a complex formed by a series of individual proteins which are encoded by four genes, namely accA, -B, -C and -D25. Those genes encode four proteins, which form a complex in vivo. Each subunit possesses different enzymatic functions. The most crucial enzyme in this step, the biotin carboxylase, is encoded by the gene accC, which catalyzes binding of a carboxyl group to biotin by consumption of adenosine triphosphate (ATP) and bicarbonate (HCO3-).
Furthermore, the accB gene encodes the Biotin Carboxyl Carrier Protein (BCCP), which is needed to transfer the carboxyl group onto the carboxyltransferase and subsequently regenerates the biotin.
AccA and accD encode the α- and β-subunits of the carboxyl transferase, which converts Acetyl-CoA to Malonyl-CoA and is subsequently converted to Malonyl-ACP by the acyl carrier protein. Malonyl-ACP is the main precursor for chain elongation - after which the elongated chain becomes the substrate of the thioesterase, which terminates the elongation and releases the free fatty acid.
This subproject started with the isolation of the accA, -B, -C and -D genes from the E. coli K12 strain and further usage of E. coli BL21 (DE3) for heterologous protein overexpression. For overexpression, the previously named genes were subsequently cloned into pET21a (Novagen). The expression is controlled by the strong T7 promoter BBa_K2406020, enabling transcription by T7 polymerase, inducible either by lactose or IPTG. As a proof of concept, the mentioned genes encoding the ACC complex were separately overexpressed and ACC-protein subunits accA and accB were purified by affinity and size exclusion chromatography. Further investigations covered the complete overexpression of the ACC complex, using an operon-like vector construct with the subsequent positioning of the single acc-genes (Fig 18). With this construct, the fatty acid content in the cells transformed and expressing the Acc complex can be measured and compared to native fatty acid composition of the producing host by gas chromatography.
Milk proteins are usually divided into two categories: Those proteins that are very insoluble and occur in cheese, the caseins, and those proteins that get filtered out during cheese making, which are called whey proteins. Caseins make up around 82% of total protein in cow’s milk26.
Whey proteins form a very diverse class of polypeptides, thus the focus in the present project is on the two most abundant ones at around 50 % and 20 % of whey protein: ɑ-lactalbumin and β-lactoglobulin. Caseins are made up of just four different proteins: ɑ-s1-, ɑ-s2-, β- and κ-casein.
The gene sequences for the milk proteins were acquired by reverse-translation of the amino acid sequence, as the genes could not been directly isolated from the native organisms. Further, the obtained gene sequences were codon-optimized for overexpression in Escherichia coli and synthesized commercially.
For the overexpression of the target genes in E. coli, the strain BL21 (DE3) was chosen as an expression host. This strain represents a very convenient model system because of its beneficial biological traits. The BL21 (DE3) strain does not express a typical set up of endogenous proteases and has the T7-Polymerase in its genome27. The genes for the milk proteins were cloned into the T7 promoter based vector, named pET22b (Novagen). The constructs contain a fused Histidine tag (6xHis) (Fig. 19) which enables the purification of overexpressed proteins by affinity chromatography and immunodetection via western blot. The T7 promoter drives the expression strongly, allowing transcription by the T7-Polymerase. The overexpression of this polymerase is inhibited by lacI and can be induced by either lactose or IPTG.

Bacillus subtilis was chosen as the primary production organism after the feedback of experts, as it can secrete proteins directly into the medium. This is advantageous for a food product, as fewer bacterial residues have to be removed during the purification. B. subtilis is better suited for this purpose as a gram positive bacterium, because it only has one cell membrane across which the protein has to be transported.
The research group of Prof. Dr. Karl-Erich Jaeger from the Institute of Molecular Enzyme Technology at Forschungszentrum Jülich provided us with plasmid variants with several different peptide signals, which facilitate secretion. The variants were all used, as the optimal peptide signal is different for each protein. Each milk protein was cloned into each plasmid variant, so the optimal peptide signal could be identified for each milk protein. They also provided the B. subtilis DB430 strain, which expresses fewer secreted proteases, increasing the stability of all secreted proteins8.
The pBSMul1 plasmid was used with the following signal peptides: sslipA, SPYurl, SPNprE, SPPel and SPEpr. In this plasmid, protein expression is controlled by the constitutive HpaII promoter (Fig. 20). pBSMul1 plasmids carry resistances to ampicillin and kanamycin. The milk proteins were expressed fused to a Histidine-tag, to enable purification by affinity chromatography and immunodetection in Western blot.

For overexpression in Synechocystis sp. PCC 6803, the pSHDY plasmid7, was used with the Pcpc560 promoter, a strong constitutive promoter from Synechocystis28. The plasmid carries a Spectinomycin and Chloramphenicol resistance. The genes were inserted by restriction-cloning downstream of the Pcpc560 promoter. The ligation product was used for transformation of E. coli DH5ɑ and this strain was used for conjugation of Synechocystis via triparental mating. Synechocystis was cultured in BG11 medium, which contains the elements and micronutrients needed for photoautotrophic growth of the cyanobacterium.

Lactoferrin is a multifunctional protein from the transferrin group, which is assigned to serine proteases due to its enzymatic activity. In addition, lactoferrin also has nuclease activity and is an iron chelator29.
Lactoferrin consists of a polypeptide chain with about 700 amino acids and forms two similar globular domains called N-lobes and C-lobes. The N lobe is formed by amino acid residues 1 to 333, the C lobe by amino acid residues 345 to 692. The two domains are connected by a short α helix30.
Each flap consists of two subdomains (N1 and N2 as well as C1 and C2) and contains one position each for binding iron and another for glycosylation. The extent of glycosylation can vary, which explains the variation in molecular weight between 76 and 80 kDa.
Lactoferrin is a basic protein. Its isoelectric point is at a pH of 8.7. It can reversibly bind two metal ions in its domains, e.g. iron, zinc or copper. The affinity of lactoferrin to iron is about 300 times higher than that of transferrin. A distinction is made between hololactoferrin rich in iron and iron-free apolactoferrin31.
The protein lactoferrin reduces inflammation factors and thus promotes the absorption of iron into the blood and its availability in the body.
The heterologous expression of proteins has an important industrial role, e.g. for the production of insulin32. Identification of new peptide and protein pharmaceuticals and the optimization of the expression of known pharmaceuticals represent a huge research sector. Around 155 pharmaceuticals and vaccines developed by bio-pharmaceutical companies were approved by the US Food and Drug Administration in 2002, the amount of approved recombinant proteins quickly rose to over 200 in 200933. Besides insulin, medically important proteins like albumin and the human growth hormone (HGH) are produced by microbes or higher organisms.
Small proteins are usually expressed in prokaryotic organisms like Escherichia coli, which enables easy, quick and cheap protein expression34. Disadvantages are the lack of post-translational modification and glycosylation, the difficult expression of large proteins and proteins with disulphide bonds34,35.
These drawbacks can be compensated by using eukaryotic yeast as an expression host. Two of the primarily used yeast expression systems are Saccharomyces cerevisiae and Pichia pastoris36.
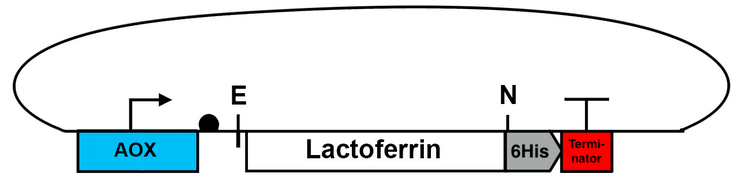
In P. pastoris the enzyme alcohol oxidase (AOX) catalyses the first reaction in the methanol utilization pathway, encoded by the two genes AOX1 and AOX2. AOX1 makes up the major enzymatic activity, the AOX1 promoter is methanol induced and therefore the AOX1 gene is highly expressed in the presence of methanol37. This leads to high recombinant protein yields of genes that are transformed into P. pastoris under influence of the AOX1 promoter38. P. pastoris expression kits are available commercially, vectors for transformation include an antibiotic resistance gene for selection and tags for protein detection and purification39.
The pPICZB Vector from the EasySelect™ Pichia Expression Kit from Thermofisher Scientific was used, which contains an inducible AOX1 promoter and a Zeocin™ resistance gene.
The gene lactoferrin originates from the organism Bos Taurus and was synthesized commercially. In addition, a NotI and an Eco72I interface were placed at both ends of the gene to clone the gene into the pPICZB vector (Fig. 22).
References
1: Steinfeld, Henning & Gerber, Pierre J. & Wassenaar, Tom & Castel, Vincent & Rosales, Mauricio & De haan, Cornelis. (2006). Livestock's Long Shadow: Environmental Issues and Options.
2: Maria Müller‐Lindenlauf, Christine Cornelius, Sven Gärtner, Guido Reinhardt, Nils Rettenmaier, Tobias Schmidt "Umweltbilanz von Milch und Milcherzeugnissen Status quo und Ableitung von Optimierungspotenzialen."
de/uploads/tx_news/IFEU-VDM-Milchbericht-Umweltbilanz-2014_01. pdf>[quoted 01.11. 2016] (2014)."
3: Bhavbhuti M. Mehta, Handbook of Food Chemistry pp 511-55
4: Wenjuan Zha, Sheryl B. Rubin-Pitela, Zengyi Shao, Huimin Zhao "Improving cellular malonyl-CoA level in Escherichia coli via metabolic engineering." Metabolic engineering 11.3 (2009)
5: Brockmeier, Ulf. "New strategies to optimize the secretion capacity for heterologous proteins in Bacillus subtilis." Biowissenschaften der Ruhr-Universitat Bochum (2006).
6: Lun Yao, Ivana Cengic, Josefine Anfelt, Elton P. Hudson Multiple gene repression in cyanobacteria using CRISPRi. ACS synthetic biology, 2015, 5. Jg., Nr. 3, S. 207-212.
7: Behle, Anna, Pia Saake, and Ilka M. Axmann. "Comparative analysis of inducible promoters in cyanobacteria." bioRxiv (2019): 757948.
8: Doi, Roy H., Sui-Lam Wong, and Fujio Kawamura. "Potential use of Bacillus subtilis for secretion and production of foreign proteins." Trends in Biotechnology 4.9 (1986): 232-235.
9: Wolk CP, Vonshak A, Kehoe P, Elhai J: Construction of shuttle vectors capable of conjugative transfer from Escherichia coli to nitrogen-fixing filamentous cyanobacteria. Proceedings of the National Academy of Sciences 2004, 81:1561–1565.
10: Thorsten Heidorn, Daniel Camsund, Hsin-Ho Huang, Pia Lindberg, Paulo Oliveira, Karin Stensjö, Peter Lindblad "Synthetic biology in cyanobacteria: engineering and analyzing novel functions." Methods in enzymology. Vol. 497. Academic Press, 2011. 539-579.
11: Fuzhong Zhang, James M Carothers, Jay D Keasling. “Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids” Nature Biotechnology volume 30, pages 354–359 (2012)
12: Marta Lubary, Gerard W. Hofland, Joop H. ter Horst ”The potential of milk fat for the synthesis of valuable derivatives” European Food Research and Technology January 2011, Volume 232, Issue 1, pp 1–8
13: Lindmark Månsson, Helena. "Fatty acids in bovine milk fat." Food & nutrition research 52.1 (2008): 1821.
14: Katayoon Dehesh, Aubrey Jones, Deborah S. Knutzon, Toni A. Voelker "Production of high levels of 8: 0 and 10: 0 fatty acids in transgenic canola by overexpression of Ch FatB2, a thioesterase cDNA from Cuphea hookeriana." The Plant Journal 9.2
15: Kamran Jawed, Anu Jose Mattam, Zia Fatma, Saima Wajid, Malik Z. Abdin, Syed Shams Yazdani "Engineered production of short chain fatty acid in Escherichia coli using fatty acid synthesis pathway." PloS one 11.7 (2016): e0160035.
16: Xiping Liu, Haiying Yu, Xu Jiang, Guomin Ai, Bo Yu, Kun Zhu "Biosynthesis of butenoic acid through fatty acid biosynthesis pathway in Escherichia coli." Applied microbiology and biotechnology 99.4 (2015): 1795-1804.
17: Lei, Anping & Chen, Huan & Shen, Guoming & Hu, Zhangli & Chen, Lei & Wang, Jiangxin. (2012). Expression of fatty acid synthesis genes and fatty acid accumulation in Haematococcus pluvialis under different stressors. Biotechnology for biofuels. 5. 18. 10.1186/1754-6834-5-18.
18: Cho, Hyeseon, and John E. Cronan. "Defective export of a periplasmic enzyme disrupts regulation of fatty acid synthesis." Journal of Biological Chemistry 270.9 (1995): 4216-4219.
19: Zinchenko, Vladislav & Piven, I.V. & Melnik, V.A. & Shestakov, S.V.. (1999). Vectors for the Complementation Analysis of Cyanobacterial Mutants. Russian Journal of Genetics. 35. 228-232.
20: Yan-Ning Zheng, Ling-Ling Li, Qiang Liu, Jian-Ming Yang, Xiang-Wei Wang, Wei Liu, Xin Xu, Hui Liu, Guang Zhao & Mo Xian "Optimization of fatty alcohol biosynthesis pathway for selectively enhanced production of C12/14 and C16/18 fatty alcohols in engineered Escherichia coli." Microbial cell factories 11.1 (2012): 65
21: VERMAAS, Wim. Molecular genetics of the cyanobacterium Synechocystis sp. PCC 6803: Principles and possible biotechnology applications. Journal of Applied Phycology, 1996, 8. Jg., Nr. 4-5, S. 263-273.
22: Toru Tobe,* Noriko Nakanishi, and Nakaba Sugimoto “Activation of Motility by Sensing Short-Chain Fatty Acids via Two Steps in a Flagellar Gene Regulatory Cascade in Enterohemorrhagic Escherichia coli” INFECTION AND IMMUNITY, Mar. 2011, p. 1016–1024
23: http://www.nutrientsreview.com/lipids/long-chain-fatty-acids-lcfa.html
24: Freiberg C, Brunner NA, Schiffer G, Lampe T, Pohlmann J, Brands M, Raabe M, Häbich D, Ziegelbauer K. "Identification and characterization of the first class of potent bacterial acetyl-CoA carboxylase inhibitors with antibacterial activity." Journal of Biological Chemistry 279.25 (2004): 26066-26073.
25: Choi-Rhee, Eunjoo, and John E. Cronan. "The biotin carboxylase-biotin carboxyl carrier protein complex of Escherichia coli acetyl-CoA carboxylase." Journal of Biological Chemistry 278.33 (2003): 30806-30812.
26: http://milkfacts.info/Milk%20Composition/Protein.htm
27: Wood, W.B. 1966. Host specificity of DNA produced by Escherichia coli: bacterial mutations affecting the restriction and modification of DNA. J.Mol.Biol. 16:118-133
28: Zhou, Jie & Zhang, Haifeng & Meng, Hengkai & Zhu, Yan & Bao, Guanhui & Zhang, Yanping & Li, Yin & Ma, Yanhe. (2014). Discovery of a super-strong promoter enables efficient production of heterologous proteins in cyanobacteria. Scientific reports. 4. 4500. 10.1038/srep04500.
29: Legrand, D., Elass, E., Carpentier, M., & Mazurier, J. (2005). Lactoferrin. Cellular and Molecular Life Sciences, 62(22), 2549.
30: Ward, P. P., Paz, E., & Conneely, O. M. (2005). Lactoferrin. Cellular and Molecular Life Sciences, 62(22), 2540.
31: Iyer, S., & Lonnerdal, B. (1993). Lactoferrin, lactoferrin receptors and iron metabolism. European journal of clinical nutrition.
32: Ebina Y, Edery M, Ellis L, Standring D, Beaudoin J, Roth RA, Rutter WJ (1985) Expression of a functional human insulin receptor from a cloned cDNA in Chinese hamster ovary cells. Proc Natl Acad Sci U S A 82: 8014–8018
33: Demain AL, Vaishnav P (2009) Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv 27: 297–306
34: Terpe K (2006) Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 72: 211–222
35: Tran A-M, Nguyen T-T, Nguyen C-T, Huynh-Thi X-M, Nguyen C-T, Trinh M-T, Tran L-T, Cartwright SP, Bill RM, Tran-Van H (2017) Pichia pastoris versus Saccharomyces cerevisiae: a case study on the recombinant production of human granulocyte- macrophage colony-stimulating factor. BMC Res Notes 10: 148
36: Cereghino JL, Cregg JM (2000) Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 24: 45–66
37: Byrne B (2015) Pichia pastoris as an expression host for membrane protein structural biology. Curr Opin Struct Biol 32: 9–17
38: Lin-Cereghino J, Wong WW, Xiong S, Giang W, Luong LT, Vu J, Johnson SD, Lin- Cereghino GP (2005) Condensed protocol for competent cell preparation and transformation of the methylotrophic yeast Pichia pastoris. Biotechniques 38: 44–48
39: Lin Cereghino J, Cregg JM (2000) Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 24: 45–66