Team:GO Paris-Saclay/Parts/A1

Nuclease_A1 : BBa_K3027000
BBa_K3027004
A protein that cleaves DNA : a deoxyribonuclease
A1 is a nuclease coming from bacteriophage T5. We have demonstrated that this biobrick can eliminate genomic DNA and thus be used to efficiently generate DNA-less chassis. To characterize the nuclease, its CDS has been inserted in a plasmid under the control of an inducible promoter. This plasmid was then used to transform bacteria which will express the nuclease for the study.
Materials and methods
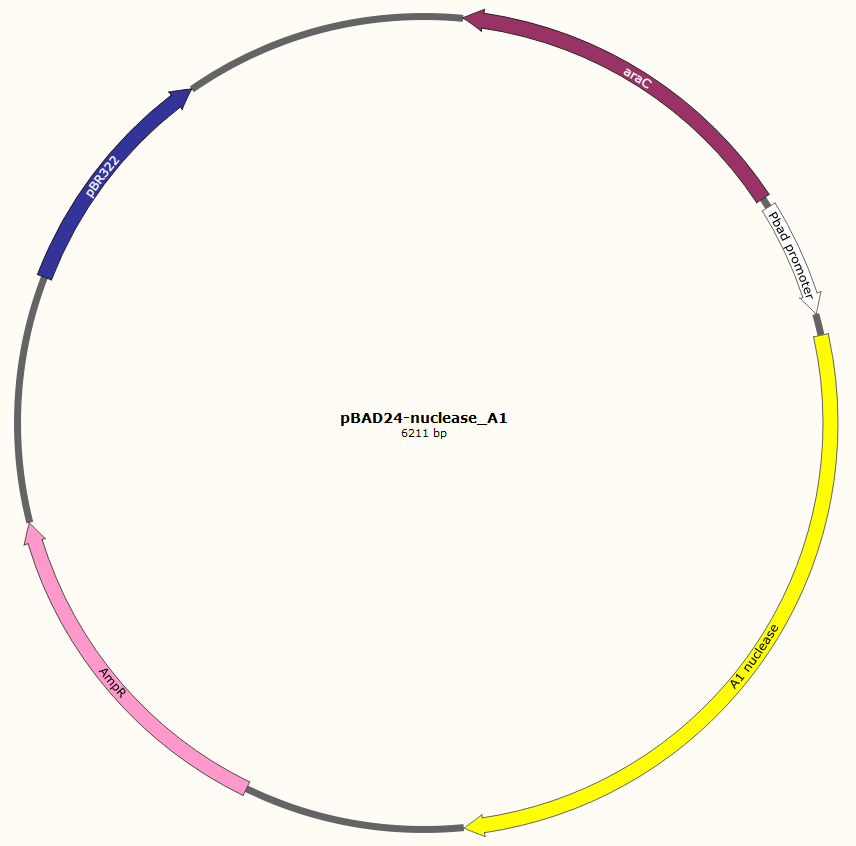 The gene encoding nuclease_A1 was cloned in the plasmid pBAD24-MoClo, resulting in pBAD24-nuclease_A1. This plasmid contains an ampicillin resistance gene, the pBR322_origin for replication, an araC gene encoding the AraC transcriptional regulator, the arabinose-inducible PBAD promoter and a RBS upstream of the nuclease gene. AraC regulates the expression of genes controlled by the PBAD. The presence of glucose in media inhibits the adenyl cyclase producing cAMP. cAMP forms a complex with the CRP protein to activate the transcription of araC gene. Therefore, when glucose is absent from media, synthesis of AraC is stimulated. This protein forms a homodimer, which represses the transcription of genes under the control of the PBAD promoter, in the absence of arabinose. Arabinose modifies the conformation of the homodimer AraC. This alteration activates the transcription of genes controlled by PBAD. This design allows a tight regulation of the inserted gene by glucose (repression) and arabinose (induction).
The gene encoding nuclease_A1 was cloned in the plasmid pBAD24-MoClo, resulting in pBAD24-nuclease_A1. This plasmid contains an ampicillin resistance gene, the pBR322_origin for replication, an araC gene encoding the AraC transcriptional regulator, the arabinose-inducible PBAD promoter and a RBS upstream of the nuclease gene. AraC regulates the expression of genes controlled by the PBAD. The presence of glucose in media inhibits the adenyl cyclase producing cAMP. cAMP forms a complex with the CRP protein to activate the transcription of araC gene. Therefore, when glucose is absent from media, synthesis of AraC is stimulated. This protein forms a homodimer, which represses the transcription of genes under the control of the PBAD promoter, in the absence of arabinose. Arabinose modifies the conformation of the homodimer AraC. This alteration activates the transcription of genes controlled by PBAD. This design allows a tight regulation of the inserted gene by glucose (repression) and arabinose (induction).
The E. coli KeioZ1F’ strain was transformed with pBAD24-nuclease_A1. The resulting strain was characterized as followed:
The strain KeioZ1F’ + pBAD24-nuclease_A1 was inoculated in 3 ml of LB supplemented with ampicillin (100 μg/mL) and glucose (0.2 %) and grown overnight at 37°C with shaking. The next day, the culture was diluted in LB supplemented with ampicillin in order to have an optical density at 600 nm (OD600) equal to 0.1. The culture was shaken at 37°C until OD600=0.4. Expression of nuclease_A1 was then induced by adding arabinose (0.2 %) to the media at time point t=0. OD600 was measured and the cell number was determined by spotting 10 μL of serial dilutions on agar plate (LB + ampicillin + glucose) at different time points: -30 min (i.e. 30 min prior to arabinose induction), +30 min (i.e. 30 min after arabinose induction), +90min, +150 min and “Day after”. This protocol was performed on at least five occasions during the summer to have a large amount of data for nuclease_A1. The negative control in all these experiments was performed with the KeioZ1F’ strain carrying pBAD24-MoClo (this plasmid does not carry the nuclease gene).

Results
Result 1: Expression of nuclease gene A1 inhibits bacterial growth and survival
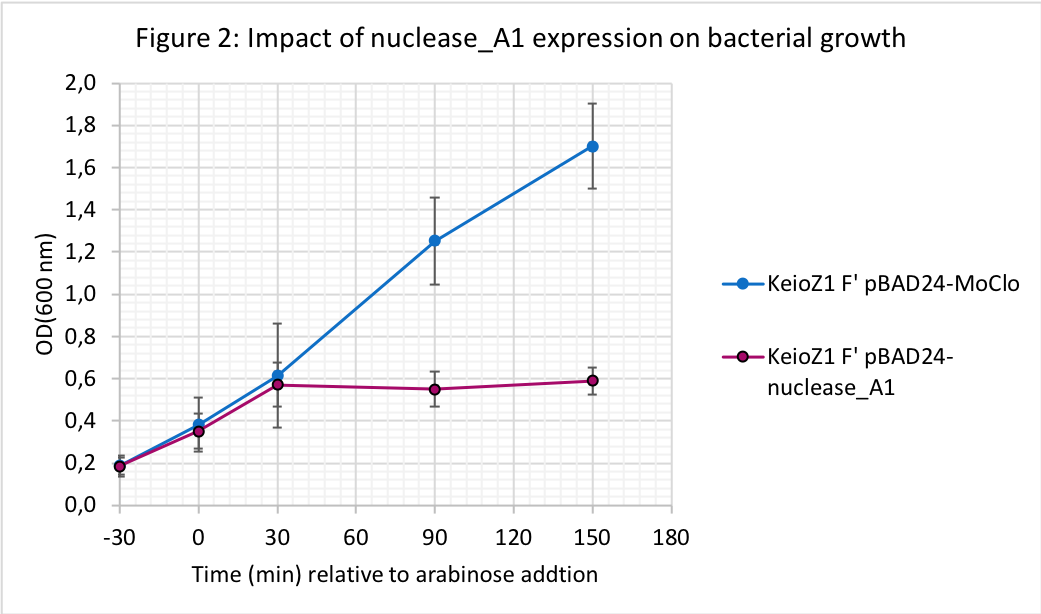
Addition of arabinose to the growth media resulted in growth arrest for the strain expressing nuclease_A1 while the OD600 kept increasing for the strain carrying the control plasmid (pBAD24-MoClo). We observed that A1 nuclease expression stopped bacterial growth until at least to 150 min upon arabinose induction.

Addition of arabinose to media did not affect cell viability of the strain with the control plasmid (Figure 3). In contrast, the viability of the strain with pBAD24-nuclease_A1 decreased ca. 500-fold within 30 min of nuclease induction by arabinose (from 3x106 CFU/mL to 6x103 CFU/mL from time -30 min to +30 min after induction). After 150min of induction of nuclease_A1 we could recover only 0.1 % of the viable bacteria that were present before induction. Therefore, A1 nuclease expression swept away up to 99.9% of the bacteria.
Result 2: Reduced genomic DNA recovery upon expression of nuclease gene A1
Next, we followed the impact of nuclease induction on bacterial genomic DNA (gDNA). Following addition of arabinose, one mL of culture was sampled at different times to extract gDNA. Figure 4A shows the results obtained using a Promega kit (cat A1120), following the manufacturer’s instructions except that RNase was not added. Figure 4B is another experiment where gDNA was extracted in the presence of RNase using a Macherey-Nagel kit . The KeioZ1F’ strain carrying pBAD24-MoClo was used as a control.

At all timepoints, gDNA and ribosomal RNA (rRNA) can be recovered from the control bacteria carrying plasmid pBAD24-MoClo. In contrast, as soon as 30 min after arabinose addition we could barely recover gDNA from cells carrying pBAD24-nuclease_A1, while rRNA was present in all samples (Figure 4A). In another experiment where RNase was added during gDNA isolation, we also observed that no gDNA could be recovered from cells expressing nuclease_A1 for 30 and 90 min (Figure 4B). Thus, induction of the expression of nuclease_A1 leads to the destruction of bacterial DNA.
Result 3: Reduced DNA staining with DAPI in cells expressing nuclease gene A1
To visualize whether cells expressing nuclease_A1 were still intact and whether they contained DNA that could be stained with DAPI, a microscopy study was performed using cell fixation and DAPI staining. Following arabinose addition, culture aliquots were taken at different time points (t = 0min, t = +30min, t = + 90min). Cells were fixed and stained according to the protocol “Fixation of cells and coloration with DAPI”. The control strain is the KeioZ1F’ strain transformed with pBAD24-MoClo.


Arabinose addition to the control cultures did not affect bacterial DNA staining with DAPI (Figure 5A). However, for those containing pBAD24-nuclease_A1, the addition of arabinose resulted in the DNA destruction in most of the bacteria already after 30 min (Figure 5B), consistent with the reduced gDNA recovery observed previously (Figure 4). After 90 min of nuclease induction only a minority of cells show some staining with DAPI. Therefore, within 30 min of expression of nuclease_A1, we generated bacterial cells free of DNA.
Result 4: Some bacterial cells survive following the expression of nuclease gene A1
When bacteria were incubated overnight with arabinose, some cells were able to survive and multiply, as witnessed by an increase in OD (Figure 6).


Therefore, some bacteria (or “cheaters”) did survive the nuclease induction. We collected two colonies from two independent induction experiments: the strains were called “Survivor 1” and “Survivor 4”. Next, we tested whether the “Survivor” strains changed phenotype compared to the original strain that was always maintained in medium with glucose (Figure 8 and 9): they no longer exhibited slowed growth or decreased survival when arabinose was added to the cells.


In order to understand how these bacteria were now able to escape death, we extracted plasmid DNA from the survivors and introduced it into strain KeioZ1F’. The resulting transformants were resistant to the negative effect of arabinose addition on growth, suggesting that the plasmids from survivors carry mutations disabling the nuclease gene or its expression. Sequence analysis of the plasmid recovered from “Survivor” 1 revealed a complete deletion of the A1 gene. For plasmid recovered from “Survivor 4” there were new DNA sequences inside the CDS of A1. These sequences were analysed with Blastx (Figure 10).

For survivor clone 4, we found a transposase inserted in the A1 CDS.
Both of the mutations explain why and how these survivors escaped death from the nuclease induction.
Result summary and perspective
Taken together, our results show that expression of our new biobrick nuclease_A1 in E. coli leads to the destruction of bacterial genomic DNA, thereby generating DNA-free cells. Loss of the nuclease gene by deletion or gene inactivation through transposase insertion are some mechanisms that can explain that 0.1% of cells escape killing. Despite these limitations, our new biobrick is a promising tool that could be used in the future for genetic containment.
